
Abstract
Background
The cysteine-aspartic acid protease caspase-3 is recognized as the main executioner of apoptosis in cells responding to specific extrinsic and intrinsic stimuli. Caspase-3 represents an interesting biomarker to evaluate treatment response, as many cancer therapies exert their effect by inducing tumour cell death. Previously developed caspase-3 PET tracers were unable to reach routine clinical use due to low tumour uptake or lack of target selectivity, which are two important requirements for effective treatment response evaluation in cancer patients. Therefore, the goal of this study was to develop and preclinically evaluate novel caspase-3-selective activity-based probes (ABPs) for apoptosis imaging.
Results
A library of caspase-3-selective ABPs was developed for tumour apoptosis detection. In a first attempt, the inhibitor Ac-DW3-KE (Ac-3Pal-Asp-βhLeu-Phe-Asp-KE) was 18F-labelled on the N-terminus to generate a radiotracer that was incapable of adequately detecting an increase in apoptosis in vivo. The inability to effectively detect active caspase-3 in vivo was likely attributable to slow binding, as demonstrated with in vitro inhibition kinetics. Hence, a second generation of caspase-3 selective ABPs was developed based on the Ac-ATS010-KE (Ac-3Pal-Asp-Phe(F5)-Phe-Asp-KE) with greatly improved binding kinetics over Ac-DW3-KE. Our probes based on Ac-ATS010-KE were made by modifying the N-terminus with 6 different linkers. All the linker modifications had limited effect on the binding kinetics, target selectivity, and pharmacokinetic profile in healthy mice. In an in vitro apoptosis model, the least hydrophilic tracer [18F]MICA-316 showed an increased uptake in apoptotic cells in comparison to the control group. Finally, [18F]MICA-316 was tested in an in vivo colorectal cancer model, where it showed a limited tumour uptake and was unable to discriminate treated tumours from the untreated group, despite demonstrating that the radiotracer was able to bind caspase-3 in complex mixtures in vitro. In contrast, the phosphatidylethanolamine (PE)-binding radiotracer [99mTc]Tc-duramycin was able to recognize the increased cell death in the disease model, making it the best performing treatment response assessment tracer developed thus far.
Conclusions
In conclusion, a novel library of caspase-3-binding PET tracers retaining similar binding kinetics as the original inhibitor was developed. The most promising tracer, [18F]MICA-316, showed an increase uptake in an in vitro apoptosis model and was able to selectively bind caspase-3 in apoptotic tumour cells. In order to distinguish therapy-responsive from non-responsive tumours, the next generation of caspase-3-selective ABPs will be developed with higher tumour accumulation and in vivo stability.
Similar content being viewed by others
Background
Programmed cell death by apoptosis is an essential biological process to eliminate unwanted or damaged cells during embryogenesis and to maintain normal tissue homeostasis (Eskandari and Eaves 2022; Morana et al. 2022). Due to the nature of the process, apoptosis is highly regulated and can proceed through two principal pathways: the intrinsic and the extrinsic pathway, both converging in the activation of the effector enzyme caspase-3, leading to cell shrinkage, chromatin condensation, cell blebbing and externalization of aminophospholipids such as phosphatidylserine (PS) and phosphatidylethanolamine (PE) and finally cell death followed by phagocytosis of the apoptotic bodies or secondary necrosis (Jiang et al. 2020; Elvas et al. 2016; Elmore 2007). Cancer is characterized by dysregulation of cell proliferation, resistance to cell death, enabling replicative immortality and angiogenesis induction, allowing unrestricted tumour growth and metastasis (Mohammad et al. 2015; Hanahan and Weinberg 2011). Therefore, many cancer treatments exert their therapeutic effect by inducing apoptosis in cancer cells, leading to a decrease of the total tumour burden (Mohammad et al. 2015). However, not all patients respond and even those who do often experience severe toxicities. Therefore, it is beneficial to evaluate the treatment response of each patient individually in order to reduce toxicity, discontinue ineffective therapy and transition to effective second line therapy earlier, and limit the need of invasive biopsies as well as minimizing the use of healthcare resources.
Currently, treatment response is evaluated by measurement of tumour volume changes by computed tomography (CT) or magnetic resonance imaging (MRI) (Eisenhauer et al. 2009; Carter et al. 2018). However, tumour volume change is a late biomarker for treatment response evaluation as it can take weeks to months before morphological changes are visible (Kummar et al. 2006). In contrast, therapy-induced tumour cell apoptosis is an early event during successful therapy. Therefore, there is a growing need to identify biomarkers associated with this process that can assess treatment response as early as possible. The assessment of apoptosis could provide an early indication of the effectiveness of cancer therapies, allowing clinicians to select the best course of treatment (Beroske et al. 2021). Multiple targets have been investigated to this purpose by positron emission tomography (PET) and single-photon emission computed tomography (SPECT) imaging, such as the externalisation of PE and PS, mitochondrial membrane collapse, DNA fragmentation and exposure, the exposure of histones and the activation of caspases (Su et al. 2021). However, caspase-3 is an interesting target as it is the main effector enzyme of apoptosis at the end of an activation cascade.
Positron emission tomography (PET) is a non-invasive technique that allows the molecular imaging of targets with high sensitivity. Several PET radiotracers targeting caspase-3 have been developed in recent years based on either caspase-3 substrates (substrate-based probes: SBPs) or inhibitors (activity-based probes: ABPs) (Nguyen et al. 2009; Chen et al. 2021, 2009, 2015, 2020; Zhou et al. 2006; Smith et al. 2008; Waldmann et al. 2015; Glaser et al. 2019; Udemba et al. 2015; Su et al. 2013a; Engel et al. 2018; Ostapchenko et al. 2019; Hight et al. 2014; Rapic et al. 2017; Elvas et al. 2019; Shen et al. 2013; Qiu et al. 2019; Lin et al. 2022a). Peptide-based SBPs are cleavable substrates of caspase-3 of which the radioactive tag is retained in the cell after cleavage. An advantage of SBPs is that an increased accumulation of activity can occur within the tumour cell due to the capability of a single enzyme to cleave multiple SBPs. Nanoaggregrate probes are SBPs that undergo intra- or extramolecular condensation reactions after cleavage of a recognition sequence, followed by the formation of large aggregates due to π-π stacking which increases cellular retention of the radiotracers (Lin et al. 2022b). ABPs, on the other hand are small-molecule or peptidic inhibitors consisting of an electrophilic warhead that covalently binds the active site of the protease, a recognition sequence that offers target selectivity, and a radioactive tag. An advantage of ABPs is that the electrophilic warhead can confer additional selectivity to the radiotracer (Vickers et al. 2014; Solania et al. 2019). Despite promising preclinical results and initial clinical studies, most relevant with [18F]ICMT-11 and [18F]CP-18, none of the caspase-3 probes have progressed to further clinical development due to low absolute tumour uptake (Nguyen et al. 2009; Su et al. 2013b). The low absolute uptake can have multiple reasons, such as, the dynamic process of caspase-3 activation, making it challenging to choose a meaningful timepoint for imaging, heterogeneous patient responses to treatment, and high apoptosis levels in healthy organs like the gut resulting in a high background signal. In addition to the previously mentioned challenges, an optimal caspase-3 radiotracer also needs to be able to enter the cells to target the intracellular caspase-3, and, have a fast clearance from the body in order to achieve a high tumour-to-background ratio (Beroske et al. 2021). A review covering the different caspase-3 tracers up to 2021 has been published by our group (Beroske et al. 2021).
A first generation of caspase-3-targeting peptidic ABPs developed by our group contained the commonly used DEVD tetrapeptide as the recognition sequence, an acyloxymethyl ketone (AOMK) warhead that binds irreversibly to the cysteine side chain of caspase-3 in a SN2-like reaction, and a 18F-labelled prosthetic group introduced through click chemistry (Scheme 1A). Although fast binding kinetics between the ABPs and caspase-3 were measured, suboptimal tumour uptake and low selectivity for caspase-3 over other proteases were observed (Rapic et al. 2017; Elvas et al. 2019).

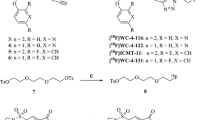
A Structure of [18F]MICA-302, a first generation caspase-3 ABP based on the DEVD recognition sequence and a AOMK warhead. B structure of Ac-DW3-KE. C Structure of Ac-ATS010-KE. The moieties that are changed between each generation are indicated in yellow
In order to optimize the selectivity for caspase-3 over related proteases, the Wolan Lab have investigated novel warheads and recognition sequences. A fourfold increase in selectivity for caspase-3 over the highly homologous caspase-7 was achieved by introducing a 5-methyl-2-thiophene carboxylate-derived ketoester group, termed KE, as a unique prime side warhead on the DEVD sequence (Vickers et al. 2013). Further development of the non-prime side recognition sequence led to the inhibitor Ac-DW3-KE (Ac-3Pal-Asp-βhLeu-Phe-Asp-KE), which showed 120-fold selectivity for caspase-3 against caspase-7 and 58-fold against caspase-8 (Scheme 1B) (Vickers et al. 2013). However, in vitro kinetic assays demonstrated that Ac-DW3-KE is a slow inhibitor that is 500 times less efficient in the inactivation of caspase-3 than Ac-DEVD-KE. By replacing the βhLeu of the DW3 sequence with a pentafluorophenylalanine residue, the Wolan Lab achieved a 154-fold increase of kinact/Ki for caspase-3 with a kinact/Ki for caspase-3 ninefold higher compared to caspase-7. This inhibitor was termed Ac-ATS010-KE (Ac-3Pal-Asp-Phe(F5)-Phe-Asp-KE) (Scheme 1C). Furthermore, elongation of the N-terminus with biotin resulted in a negligible effect on binding kinetics, which was expected as residues beyond P5 lie outside the active site of caspase-3. Finally, Ac-ATS010-KE was able to protect Jurkat cells from MegaFasL-induced apoptosis, and interestingly, it was shown that per-methylated compounds perform poorly when compared with non-methylated compounds in a cell viability assay, rendering per-methylation for cell uptake obsolete and bypassing the required hydrolysis of the methyl ester groups (Solania et al. 2019).
In this work, a small library of novel radiotracers based on the Ac-DW3-KE and Ac-ATS-010-KE inhibitors was developed and evaluated in vitro and in vivo. Different linkers were inserted between the N-terminus of the peptides and the radiolabel, including hydrocarbon, amino acid, polyethylene glycol (PEG) and carbohydrate linkers. This allowed the modulation of probe charge and lipophilicity, ultimately affecting probe uptake and pharmacokinetics.
Methods
General
All chemicals, reagents, and solvents (laboratory grade) were acquired from commercial suppliers (Sigma-Aldrich, Acros, TCI-Europe, FluoroChem and BioRad) and used without further purification. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance DRX 400 MHz spectrometer and data were analysed with MestReNova analytical chemistry software. 1H and 13C spectra were referenced to residual solvent peaks, and coupling constants are given in hertz. High resolution mass spectrometry (HRMS) analysis was performed using a Q-TOF II instrument (Waters, Manchester, U.K.). UPLC-MS analyses were performed on a Waters Acquity UPLC system coupled to a Waters TQD ESI mass spectrometer and TUV detector. Chromatographic purifications were performed with a Biotage ISOLERA One flash system equipped with an internal variable dual-wavelength diode array detector (200 − 400 nm). Reverse-phase high performance liquid chromatography (HPLC) was performed on a Waters 2545 HPLC equipped with a Waters 2998 diode array detector, a Micromass Quattro microTM model and a Waters 2767 fraction collector. For the purification of the final compounds, a Waters Xbridge BEH C18 OBD 19 mm × 100 mm (5 µm) column, with a gradient of water + 0.1% formic acid (FA) (mobile phase A) and acetonitrile (MeCN) + 0.1% FA (mobile phase B) at a flow rate of 19 mL/min was used. HPLC analysis was performed on a Shimadzu LC20 HPLC system (Shimadzu, Kyoto, Japan) coupled to a SPD-20AV UV–VIS spectrophotometric detector (Shimadzu, Kyoto, Japan) and a NaI-detector 2 × 2″ pinhole (Raytest, Straubenhardt, Germany) and Gabi Star isotope measuring system (Raytest, Straubenhardt, Germany) and on a LC2040C-3D plus (Shimadzu, Kyoto, Japan) coupled to a LCMS-2020 series (Shimadzu, Kyoto, Japan) and a GABI Nova radio-flow monitor (Elysia S.A., Angleur, Belgium).
Biochemical inhibition assay
Inhibition studies of 4–8 were performed as previously described by Solania et al. (2019) Briefly, inhibitors, substrates, and enzymes were diluted into a caspase-3 assay buffer containing 20 mM PIPES, pH 7.4, 10 mM NaCl, 1 mM EDTA, 10 mM DTT, and 10% sucrose. Solutions used to assay casp-9 and casp-10 were supplemented with 0.7 M sodium citrate. A 20 μL (2.5 ×) mixture of corresponding 2-aminoacridin-9(10H)-one (AMAC) substrate (final concentration 100 μM; Ac-DEVD-AMAC for casp-3 and casp-7, Ac-VEID-AMAC for casp-6, and Ac-IETD-AMAC for casp-8, -9, and -10) and inhibitor were dispensed into a black 96-well Costar flatbottom polystyrene plate. Then, a 60 μL (1.67 ×) enzyme solution (final concentration: 1 nM casp-3, 25 nM casp-6, 5 nM casp-7, 25 nM casp-8, 100 nM casp-9, and 20 nM casp-10) was added, mixed for 10 s at 1000 rpm, and the fluorescence was immediately read on a PerkinElmer EnVision plate reader with a λex = [320] and λem = [460]. Data were analysed using GraphPad Prism to determine kinact and Ki. However, progress curves were fit to F = -[v0(1-exp(-kobst))/kobs-Fmax] to account for the loss in fluorescence of AMAC substrates.
Due to the unavailability of corresponding AMAC substrates, the kinact/Ki of 9 was measured using commercially available Ac-DEVD-AMC (ENZO life sciences) and the procedure was performed accordingly with commercially acquired caspase-3 and caspase-7 (ENZO life sciences). In brief, 9, reference inhibitor Ac-ATS010-KE, Ac-DEVD-AMC, and enzymes were diluted in the previously described caspase-3 assay buffer. A 10 µL solution of Ac-DEVD-AMC (final concentration 50 µM) and inhibitor (final concentration 20 to 80 nM) were dispensed in a Greiner polystyrene 96 well, half-area, transparent plate. Then, 30 µL of a 1.67 nM solution of caspase-3 (final concentration 1 nM) was added, mixed for 3 s at 1000 rpm and immediately read on a TECAN infinite M200 plate reader with λex = [360] and λem = [460]. To account for a small background activity that was observed, the data were fit to F = F0 + (v0-vs)*(1—exp(-kobs*t))/kobs + vs*t and analysed using Grafit 7 to calculate the kobs. kinact/Ki was determined from the slope of the linear range of the kobs vs [I] curve.
Tracer synthesis
[18F]MICA-314, [18F]MICA-315, [18F]MICA-316, [18F]MICA-317 and [18F]MICA-318 synthesis
No carrier-added [18F]F– was produced in a Siemens Eclips HP cyclotron by bombardment of an enriched [18O]water target with a 50 µA proton beam for 20 min. The resulting [18F]F−/H2[18O]O was transferred to a TRASIS AllInOne PET tracer synthesizer and passed through a preconditioned QMA Sep-Pak Light cartridge to capture [18F]F−. The fixed [18F]F− was eluted from the QMA cartridge with a solution of 0.75 mL Kryptofix2,2,2 0.07 M/K2CO3 0.03 M in 1 mL MeCN:water 95:5 (v/v) and collected in a reaction vial from which the solvents were removed under a stream of helium (0.5 bar) at 90 °C for 4.5 min and 130 °C for 6.5 min. 20 mg of the tosylate precursor (1-methyl-4-(pent-4-yn-1-ylsulfonyl)benzene dissolved in 500 µL anhydrous MeCN was added to the anhydrous [18F]fluoride and reacted at approximately 100 °C for 10 min.
The resultant [18F]5-Fluoro-pentyne was distilled at 130 °C for 3 min into a second reaction vessel containing the click reaction mixture (2 mg of the corresponding azide precursors (15–19) in the presence of 75 µL 0.1 M aqueous copper sulfate, 10 mg sodium ascorbate, and 3.75 mg TBTA dissolved in a minimal amount of DMF) and left to react for 20 min at room temperature. The reaction mixture was purified via semi prep purification on a LUNA C18 (2) 100A, 250 × 10 mm (SI figures 7–11). The collected fraction was diluted to < 10% organic solvent with water, trapped on a Sep-Pak C18 Plus Light Cartridge, washed with 5 mL of water and eluted with 0.5 mL of EtOH. Finally, the eluate was diluted with 4.5 mL 0.9% NaCl solution and the tracer solution was sterile filtered. Tracer identities were determined via HPLC analysis and radiochemical purity (RCP) was determined by calculating the AUC of the intact tracer over the total area under the curve (AUC) of the radio chromatogram (SI figures 17–21).
[18F]MICA-320 synthesis
No carrier-added [18F]F– was produced in a Siemens Eclipse HP cyclotron by bombardment of an enriched [18O]water target with a 50 µA proton beam for 20 min. The resulting [18F]F−/H2[18O]O was transferred to a TRASIS AllInOne PET tracer synthesizer and passed through a preconditioned QMA Sep-Pak Light cartridge to capture [18F]F−. The fixed [18F]F− was eluted from the QMA cartridge with a solution of 0.8 mL 7.8 mM Tetraethylammonium bicarbonate (TEAB) in MeCN:water 95:5 (v/v) and collected in a reaction vial from which the solvents were removed under a stream of helium (0.5 bar) and a gentle vacuum at 115 °C for 1.3 min, 125 °C for 3 min, 110 °C for 2.67 min and 95 °C for 2 min. 5 mg of tosylated precursor 24 dissolved in 500 µL anhydrous MeCN was added to the anhydrous [18F]fluoride and reacted at approximately 100 °C for 10 min. The reaction vial was cooled down to 40 °C and 2 mL of 10% MeCN in H2O was added for transfer. The reaction mixture was removed from the vial and diluted further with 8.5 mL of water and transferred to HPLC for semi-prep purification (SI figure 12). After collection, the fraction was diluted with 45 mL water and passed through an Sep-Pak C18 Plus Light Cartridge and the cartridge was rinsed with 5 mL water. Deprotection was performed using 1 mL of 2 M NaOH for 1 min followed by 1 mL of water. The fraction was collected in a vial containing 150 µL of acetic acid before transfer to the click reaction vial. The click reaction was performed for 20 min in a vial containing 5 mg of sodium ascorbate, 37.5 µL 0.1 M of aqueous CuSO4 solution, 1 mg THPTA and 1 mg of 20 at room temperature. The click reaction mixture was diluted with 8 mL of water before transfer to HPLC for semi-prep purification (SI figure 13). The collected fraction was diluted to < 10% MeCN with water, trapped on a Sep-Pak C18 Plus Light Cartridge and washed with 5 mL water. The tracer was eluted with 500 µL EtOH, diluted with 4.5 mL 0.9% NaCl solution and filtered sterile to generate the final formulation.
Tracer identity was determined via HPLC analysis and RCP was determined by calculating the AUC of the intact tracer over the total AUC of the radio chromatogram (SI figure 22).
[99mTc]Tc-Duramycin synthesis
[99mTc]Tc-Duramycin was prepared as previously described (Elvas et al. 2016).
[18F]MICA-304 and [18F]MICA-309 synthesis
No carrier-added [18F]F– was produced in a Siemens Eclips HP cyclotron by bombardment of an enriched [18O]water target with a 50 µA proton beam for 20 min. The resulting [18F]F−/H2[18O]O was transferred to a FLUORSYNTHON III (Veenstra instruments) PET tracer synthesizer and passed through a preconditioned QMA Sep-Pak Light cartridge to capture [18F]F−. The fixed [18F]F− was eluted from the QMA cartridge with a solution of 0.75 mL Kryptofix 222 0.029 M/K2CO3 0.014 M in MeCN: water 95:5 and collected in a reaction vial from which the solvents were removed under a stream of helium (0.5 bar) at 90 °C for 4.5 min and 130 °C for 6.5 min. Azidoethyl tosylate precursor (2 mg) dissolved in anhydrous MeCN (500 µL) was added to the anhydrous [18F]fluoride and left to react at approximately 100 °C for 6 min.
The resultant [18F]FEA was distilled at 130 °C into a second reaction vessel containing the click reaction mixture of 29.9 µmol ascorbic acid dissolved in 133 µL 0.1 M aqueous CuSO4 to which 6.6 µmol Bathophenanthrolinedisulfonic acid disodium salt hydrate (BPDS) and ~ 2 µmol of the respective alkyne precursor (15 or 16) dissolved in a minimal amount of DMF were added and left to react for 10 min at room temperature (SI Scheme 1). The reaction mixture was purified via semi-prep purification on a LUNA C18 (2) 100A, 250 × 10 mm (SI figures 5, 6). The collected fraction was diluted to 10% EtOH with 0.9% aqueous NaCl solution. Tracer identities were determined via HPLC analysis and RCP was determined by calculating the AUC of the intact tracer over the total AUC of the radio chromatogram (SI figures 14–16).
Partition coefficient determination
The partition coefficient of the radiotracers was measured using the ‘shake-flask’ method. Approximately 74 kBq of tracer was added to a test tube containing 2 mL PBS (0.01 M, pH 7.4) and 2 mL of n-octanol. The tube was thoroughly shaken, vortexed for 2 min and centrifuged at 3,000 rpm for 10 min. After separation of the layers, a 0.5 mL aliquot of each layer was transferred into two separate, pre-weighed tubes that were counted for radioactivity in an automatic gamma-counter (Wizard2 2480, Perkin Elmer). The counted activities were corrected for mass and density before calculating the octanol–water partition coefficients by dividing the radioactivity in octanol by the radioactivity in PBS from which the log10 was calculated. This experiment was performed in triplicate for each probe. The logD values are reported as mean ± SD.
In vitro mouse plasma stability determination
The stability in mouse plasma was determined in vitro by HPLC analysis of aliquots at different timepoints for a total of 2 h. Approximately 185 kBq of formulated radiotracer, [18F]MICA-314 to-320, was added to 100 µL of mouse plasma at 37 °C, which was left to shake at 300 rpm for 15, 30, 60 and 120 min. At the designated timepoints, the plasma was added to 100 µL ice-cold MeCN, vortexed for 30 s and left to rest on ice for 3 min, whereafter it was centrifuged for 7 min at 4500 rpm and 4 °C. After separation of the layers, the supernatant was transferred to a different vial and the radioactivity extraction was determined by measuring the radioactivity counts of both the supernatant and the precipitate, ranging from 89.60 ± 2.44% to 94.03 ± 1.83%. Finally, 100 µL of the supernatant was diluted 1:1 in water and injected on HPLC (SI figures 23–30) and the percentage of intact tracer per timepoint was calculated by dividing the radioactivity counts of the tracer over the radioactivity counts of the total run. Mouse plasma stability determination was performed in triplicate for each tracer. The stability of the tracer in formulation was assessed at RT up to 2 h in similar fashion.
Cell uptake in apoptotic cells
HeLa (ATCC CCL-2) human cervical cancer cells and caspase-3 deficient MCF-7 (ATCC HTB-22) human breast cancer cells were seeded in 12-well plates at a density of 0.3 × 106 cells per well, and cultured in complete DMEM medium supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 10% fetal bovine serum and 100 U/mL penicillin plus 100 mg/mL streptomycin (Life Technologies), at 37 °C under a 5% CO2 humidified atmosphere. Twenty-four hours later, the media was refreshed and the cells were treated with staurosporine (STS; 1 µM for HeLa, and 5 µM for MCF-7 cells) for up to 4 h, with or without pre-incubation with 25 µM Ac-ATS010-KE for 1 h, or 0.2% DMSO (N = 5 for each condition) (Elvas et al. 2019). Four hours after treatment, approximately 74 kBq of tracer ([18F]MICA-316 or [18F]MICA-320) was added to the wells and incubated for 1 h. To determine the cell uptake, the medium was removed, the cells were washed twice with cold PBS and the cells were collected from the wells. The radioactivity in the cells, the medium and the washed fractions were measured in an automatic gamma-counter. Cells were centrifuged at 1,000 × g for 2 min, and the pellet was washed twice with ice cold PBS. After the last centrifugation step, the pellet was resuspended in 40 µL of hypotonic lysis buffer (50 mM PIPES, 10 mM KCl, 5 mM MgCl2, 2 mM disodium EDTA, 1% (v/v) Nonidet P-40 (NP-40), 2 mM DTT, pH 7.4) and incubated for 10 min on ice, as described before (Elvas et al. 2019). Cell lysates were, passed twice through a 29G needle, centrifuged at 7000×g for 15 min at 4 °C, and the supernatant was then transferred to a fresh tube. The total protein concentration of each sample was quantified using the bicinchoninic acid (BCA) protein assay (Thermo Scientific). The cellular uptake was described as %ID/g protein.
In vivo small-animal PET imaging
For in vivo PET biodistribution studies, healthy female CD1−/− nude mice and xenograft models (6–8 weeks old, 20-25 g) were anesthetized using isoflurane (5% for induction, 2% for maintenance), placed on the animal bed in the scanner and injected via lateral tail vein catheterization with approximately 18.5 MBq of tracer (n = 3). Dynamic whole-body PET images were acquired during 60 min (12 × 10 s, 3 × 20 s, 3 × 30 s, 3 × 60 s, 3 × 150 s and 9 × 300 s frames) using an Inveon small-animal PET/CT scanner (Siemens). Following each PET acquisition, a whole-body CT scan was acquired to obtain anatomic information for segmentation. Throughout the entire PET/CT scanning procedure, the mice were maintained at constant body temperature by using a heating pad. All images were reconstructed in 8 iterations and 16 subsets using the 3D ordered subset expectation maximization (OSEM3D) algorithm utilizing a list-mode iterative reconstruction with proprietary spatially variant resolution modelling for quantitative analysis (Miranda et al. 2020). Corrections for normalization, dead time, and CT-based attenuation correction were applied. PET image frames were reconstructed using 0.776 × 0.776 × 0.796 mm3 voxels on a 128 × 128 × 159 grid. Volumes of interest (VOIs) were manually drawn on the PET/CT images using PMOD (version 3.6; PMOD Technologies, Zurich, Switzerland) to delineate the regions with distinct PET time activity pattern: heart, liver, small intestines, muscle, bone and bladder. The average organ or tissue activity per volume was obtained from the co-registered PET images and the decay-corrected time-activity curves (TACs) were extracted for each target organ. For an absolute measure of tracer uptake, normalized images were scaled according to the percentage injected dose (%ID/mL = tissue uptake [kBq/mL]/injected dose [kBq] × 100).
In vivo metabolite analysis
The blood from the above ex vivo biodistribution studies was collected in EDTA-coated tubes and used to evaluate the in vivo plasma stability of the tracers at 15, 30, 60 and 120 min (N = 3 per time point). The plasma fraction was obtained by centrifugation at 4,000 g for 7 min, and the samples were analysed as described for the in vitro mouse plasma stability determination.
Animal models
Experimental procedures and protocols were performed in accordance with European Directive 86/609/EEC Welfare and Treatment of Animals and were approved by the local ethical committee (2018–45, University of Antwerp, Belgium). All animals were housed under environmentally controlled conditions (12 h light/dark cycle, 20–24 °C, and 40–70% relative humidity) in individually ventilated cages with food and water ad libitum. Animals were assigned to experimental groups using simple randomization.
Treatment-sensitive COLO205 (ATCC-CCL-22) and treatment-resistant HT29 (ATCC-HTB-38) human colorectal cancer cells were grown in complete RPMI1640 or DMEM, respectively, supplemented with 2 mM L-glutamine, 10% fetal bovine serum, 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 mg/mL streptomycin (Life Technologies). The cell lines were cultured at 37 °C in a humidified atmosphere with 5% CO2. The xenograft models were generated by subcutaneous injection of COLO205 (2 × 106 cells in 100 μL of sterile saline) or HT29 (10 × 106 cells in 100 μL sterile saline) tumour cells into the hind leg of 6- to 8-wk-old female CD1−/− nude mice (body weight, 20–25 g; Charles River Laboratories). Tumour volumes were measured 3 times per week by using a digital calliper, with tumour volumes calculated according to the formula: (length x width2)/2. When tumours reached approximately 200 mm3, mice were treated with a single dose of conatumumab (1.5 mg/kg intraperitoneally; Amgen, Inc.; N = 5) or with the corresponding IgG1 isotype control (1.5 mg/kg intraperitoneally; Amgen, Inc.; N = 5). Mice were subjected to µPET/CT imaging before (baseline) and 24 h after therapy for treatment response assessment.
In vivo imaging of treatment induced tumour cell death
The treatment-induced tumour changes in the tumour-bearing mice were assessed with [18F]MICA-316 (n = 8) PET imaging at baseline and 24 h post-treatment. Imaging and reconstruction were performed as described above. In addition, a static whole-body PET/CT image was acquired at 120 min post-radiotracer injection (p.i.) (1 × 1200 s frame). VOIs were manually drawn on the PET/CT images to delineate tumour, heart, and muscle. Voxel-based analysis was performed by extracting the relative activity of each voxel from the tumour VOIs of the 1 h dynamic scan. A histogram of all voxel activities was calculated and the histogram of relative voxel intensities of the tumour VOIs were plotted in a box plot showing the quartiles, P5, and P95 in Graphpad Prism 10 (version 10.1.1). P95 values were compared between groups to assess the value of the high-intensity voxels, as described by Nguyen et al. (2009).
Furthermore, COLO205 xenografts (N = 2) were injected with 37 MBq of [99mTc]Tc-duramycin (reference apoptosis radiotracer) 20 h after administration of conatumumab or the IgG1 control. Four hours p.i., the animals were scanned as described before (Elvas et al. 2017), after which the animals were injected with [18F]MICA-316 and scanned as described above (Elvas et al. 2016). In brief, static whole-body single photon emission computed tomography (SPECT) imaging was performed using a µSPECT/CT collimator (VECTor/CT, MILabs) equipped with 75 pinholes of 1 mm, followed by CT acquisition (45 kV and 615 μA) for attenuation correction and anatomical coregistration with SPECT images. Mice were anesthetized through isoflurane inhalation (5% for induction and 2% for maintenance) and kept at constant body temperature during the scans. SPECT images were reconstructed with ordered-subsets expectation maximization (10 iterations, 16 subsets) and 1.2 mm3 voxel size, and smoothed with a three-dimensional isotropic Gaussian filter of 1 mm for visualization purposes. A 20% energy window centered at 140 keV photopeak was used.
Ex vivo biodistribution studies
Biodistribution studies were performed in healthy and tumour-bearing mice. Healthy female CD1−/− nude mice (6–8 weeks old, 20–25 g) were injected with 18.5 MBq of tracer (n = 12) via the lateral tail vein. At 15, 30, 60, or 120 min (n = 3 for each time point) p.i., blood was collected through cardiac puncture, and the mice were euthanized by cervical dislocation. The organs and tissues were harvested, weighed and the radioactivity in the samples was measured using an automatic γ-counter. For COLO205 (N = 3/group) and HT29 (N = 5/group) tumour xenografts, the mice were euthanized by cervical dislocation 150 min after administration of [18F]MICA-316. The uptake levels of the tracers were expressed as percentage of the injected dose per gram tissue (%ID/g tissue).
Immunohistochemistry
Tumours were fixed overnight in 10% buffered formalin and transferred to 70% ethanol until paraffin embedding. Adjacent tumour sections (5 µm) were collected at regular intervals across the whole tumour for histological analysis of apoptosis. Immunohistochemistry (IHC) for cleaved caspase-3 (CC3, rabbit monoclonal anti-CC3, 1:300 dilution, Cell Signaling Technology) was carried out according to the manufacturer’s instructions. To counterstain the sections, Mayer’s hematoxylin was used. Light microscopic images were acquired using a AxioScope A1 microscope (Zeiss) at a 20X magnification coupled with an Olympus UC30 colour camera.
Activity-based labelling of tumour homogenates with [18F]MICA-316
The tumour homogenates (20–60 µg) were incubated with approximately 185 kBq of [18F]MICA-316 for 60 min at 37 °C, in 40 µL of caspase assay buffer. The reaction was quenched by adding reducing 4X Bolt LDS sample buffer, directly loaded in Bolt 12% Bis–Tris Plus gel (Thermo Scientific), and separated by SDS-PAGE according to the vendor’s protocol. A probe alone control was run in parallel to account for any background bands that may appear in the gel. In addition, to validate selectivity of the probes a caspase inhibitor (25 µM Ac ATS010-KE) was added 30 min prior the incubation with the probes. After washing, the gels were exposed overnight to phosphor screen pates (Fujifilm). The exposed plates were imaged in a phosphor imager system (FLA7000, GE Healthcare). The digital images were analysed using ImageJ software (version 1.51; NIH) and transformed uniformly to produce images to facilitate visualization of the protein bands. The regions containing free probe in the bottom of the gels were cropped from the gel images to improve signal to background ratio. No additional bands than the ones shown were observed.
Preparation of cell lysates
Cell lysates of HeLa and MCF7 cells treated with STS or DMSO as control were prepared as described above and stored at − 80 °C for further use.
Caspase activity assay using a fluorogenic substrate in organ homogenates and cell lysates
Equal amounts of organ homogenate or cell lysate protein (20 µg) were transferred to each well of a black 96-well low-binding microplate containing 50 µM of the fluorogenic substrate, Ac-DEVD-AMC, in 100 µL of caspase assay buffer. The reaction mixture was incubated in the microplate for 1 h at 37 °C. The amount of liberated fluorescent group amino-4-methylcoumarin (AMC) was measured on a Synergy Mx microplate reader (BioTek) with excitation wavelength of 360 nm and emission detection at 460 nm. The time of incubation and the amount of substrate concentration used were previously determined to be in the linear range of substrate utilization. Based on the standard curve prepared from fluorescence readings using free AMC, the data for caspase activity represent picomoles of liberated AMC and were expressed as mean ± SD. The reported values represent the mean of three independent measurements.
Data analysis
Data were expressed as mean ± SD. Statistical analysis were performed using GraphPad Prism 10.1.1. Statistical significance between multiple datasets (not assuming normally distributed data) was assessed using the Kruskal–Wallis test followed by Dunn’s multiple comparison test. Statistical differences between two groups were assessed using the Mann–Whitney test. Differences between groups were considered statistically significant if the p-value was less than 0.05. *Indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, and **** indicates p < 0.0001.
Results
Tracer design
To overcome selectivity issues encountered in the first DEVD-based generation of caspase-3 ABPs, the design of the 2nd generation probes incorporated the DW3 sequence (Ac-3Pal-Asp-βhLeu-Phe-Asp), as this structure had shown to have a high selectivity for caspase-3. A KE warhead was introduced for 1, and binding kinetics were compared to 3, in which an AOMK warhead was used (Scheme 2A). Although 3 had faster binding kinetics, 1 was developed further due to the higher selectivity for caspase-3 compared to caspase-6 and caspase-7 (Table 1). Furthermore, the effect of sidechain methylation of Ac-DW3-KE on caspase inhibition was assessed in an in vitro cell assay, indicating that the effect was limited and methylation was not necessary for cell uptake (SI figure 1). Subsequently, 18F was introduced through copper-catalysed azide alkyne cycloaddition (CuAAC) using 2-[18F]fluoroethyl azide prosthetic group, at the N-terminus of the per-methylated peptide to generate [18F]MICA-309 (SI Scheme 1). The use of a prosthetic group labelling approach was preferred to direct labelling to avoid exposure of the peptides to harsh reaction conditions during radiosynthesis. However, low total tumour uptake in the xenograft model was observed (SI figures 2–4), hindering further investigation of [18F]MICA-309.

A Structures of 2nd generation DW3-based probes. B Structure of 3rd generation caspase-3-selective, ATS010-KE-based ABPs
As it was hypothesized that the low uptake was due to the slow binding kinetics of the DW3-KE scaffold, 3rd generation caspase-3-selective ABPs were developed based on the Ac-ATS010-KE (Ac-3Pal-Asp-Phe(F5)-Phe-Asp-KE) inhibitor which has faster binding kinetics than Ac-DW3-KE. A library of 6 ABPs (4–9) was developed by modifying the linker between the recognition sequence and the radionuclide (Scheme 2B). Similarly to [18F]MICA-309, 18F was introduced through CuAAC using radiolabelled prosthetic groups (5-[18F]fluoro-1-pentyne for [18F]MICA-314 to -318 and [18F](2R,3R,4S,5S,6S)-2-Azido-6-(fluoromethyl)tetrahydro-2H-pyran-3,4,5-triol for [18F]MICA-320). The side chains at P1 and P4 positions of the 3rd generation probes were non-methylated, as Solania and colleagues had proven that the non-methylated Ac-ATS010-KE performed better than the methylated Ac-ATS010-KE in an in vitro viability assay (Solania et al. 2019).
In vitro kinetic assays
Biochemical inhibition experiments demonstrated that all peptide inhibitors derived from Ac-ATS010-KE (4 – 8) inhibit caspase-3 with a remarkably high inactivation rate (kinact/Ki ∼ 500,000 – 1,600,000 s−1 M−1), and that the most selective inhibitor, 7, has a 14-times higher inactivation rate on caspase-3 as compared to caspase-7 (Table 1). Elucidating that the N-terminal modifications of Ac-ATS010-KE had minimal impact on binding kinetics to caspase-3, and the selectivity over the other caspases.
The addition of a carbohydrate on the N-terminus led to a more pronounced decrease in binding kinetics to caspase-3 for 9 (5.19 ± 0.44 × 104.s−1.M−1) in comparison to the reference inhibitor Ac-ATS010-KE (46.48 ± 9.96 × 104.s−1.M−1). However, it is important to note that instead of using Ac-DEVD-AMAC as a substrate, Ac-DEVD-AMC was used to measure the binding kinetics of 9 alongside the original inhibitor Ac-ATS010-KE, which may account for a decrease in rate.
Radiochemistry
The radiotracers [18F]MICA-314, [18F]MICA-315, [18F]MICA-316, [18F]MICA-317 and [18F]MICA-318 (Scheme 3A) were obtained by CuAAC of 5-[18F]fluoro-1-pentyne, prepared by nucleophilic substitution of tosylated precursor with [18F]F−, with the corresponding azide precursors (15–19). The tracers were obtained in sufficient radiochemical yields and high radiochemical purities for in vitro and in vivo investigations (Table 2).

A Radiosynthesis of [18F]MICA-314 to -318. B Radiosynthesis of [18F]MICA-320
[18F]MICA-320 (Scheme 3B) was obtained by CuAAC reaction of the alkyne precursor of 20 with [18F](2R,3R,4S,5S,6S)-2-Azido-6-(fluoromethyl)tetrahydro-2H-pyran-3,4,5-triol, which was obtained by nucleophilic substitution of 24 with [18F]F−, following a previously reported protocol (Stéen et al. 2021). The tracer was obtained in sufficient decay-corrected to end of bombardment (EOB) radiochemical yield (7.9 ± 4.4%) and high radiochemical purity (98.9 ± 0.2%) (Table 2). The molar activities (Am) of both [18F]MICA-316 and [18F]MICA-320 were in the range of 50–300 GBq/µmol at end of synthesis (EOS).
The partition coefficients, expressed as LogD values, of the tracers were determined experimentally and ranged from -2.58 ± 0.01 to -0.42 ± 0.18, indicating that the six tracers were hydrophilic. As expected the Glutamic acid (E) in the side chain of [18F]MICA-314 made the tracer more hydrophilic whereas the uncharged 6-aminohexanoicacid (Acp) linker made [18F]MICA-316 less hydrophilic (Table 2).
Stability
After radiosynthesis, the stability of the tracers in formulation was assessed up to 2 h at RT to determine if the tracer remained stable for the duration of the experiments. All the radiotracers showed excellent stability in formulation over 2 h. Next, the stability was investigated in mouse plasma to determine if the tracer had potential for in vivo use. A modest degradation of the tracers [18F]MICA-314, -316, -317, -318 and -320 was observed in mouse plasma at 37 °C, with 77–88% of intact tracer remaining after 2 h for [18F]MICA-314,-316, -317, -318 and -320. However, [18F]MICA-315 showed a more pronounced degradation, with only 58.4 ± 0.6% of intact tracer remaining after 2 h (Fig. 1).

Radiotracer stability in formulation (blue, circle, N = 1), mouse plasma (red, square, N = 3) and in vivo in CD1−/− nude mice (green, triangle, N = 3) of [18F]MICA-314, [18F]MICA-315, [18F]MICA-316, [18F]MICA-317, [18F]MICA-318 and [18F]MICA-320
In vivo, the tracers showed a faster degradation in serum, with less than 30% of tracer remaining intact for [18F]MICA-314 to -318, and, 58.8 ± 22.5 intact [18F]MICA-320 after 30 min in healthy CD1−/− nude mice. Similar to previously developed radiolabelled peptides, the in vitro and in vivo degradation resulted in the formation of polar metabolites eluting earlier when using reverse phase HPLC. It has been hypothesized that the peptide backbone undergoes enzymatic degradation leading to peptide hydrolysis with the formation of polar radiometabolites; however, the identity of the metabolites was not confirmed experimentally. Interestingly, [18F]MICA-320, the tracer with a carbohydrate-derived prosthetic group, demonstrated a higher stability in vivo after 30 min than the other tracers. However, the difference with [18F]MICA-317 was not significant.
Biodistribution study
To increase target-to-background ratio, it is important that a radiotracer demonstrates good pharmacokinetic (PK) properties: fast clearance from the blood and non-target tissues. To gain insight into the PK profile of the developed caspase-3-targeting ABPs, in vivo µPET imaging and ex vivo biodistribution data were acquired. Figure 2 shows the time activity curves of the heart, muscle, bone and excretory organs: liver, small intestines and bladder up to 60 min p.i. in healthy CD1−/− nude mice. The 6 radiotracers were characterized by an initial peak in radioactivity in the heart, followed by a rapid blood clearance. All radiotracers showed a mixed renal-hepatobiliary clearance with an early peak in the liver and high activity in the bladder and small intestines up to 60 min. Furthermore, the lack of uptake in the bone for the different radiotracers showed that in vivo defluorination was negligible. The lack of non-specific uptake in the other organs resulted in a low background, making the radiotracers suitable for apoptosis imaging within 60 min p.i. The ex vivo biodistribution performed at 15, 30, 60 and 120 min p.i. validated these results (SI figure 31).

TAC and maximum intensity projection (30–60 min) of [18F]MICA-314, [18F]MICA-315, [18F]MICA-316, [18F]MICA-317, [18F]MICA-318, and [18F]MICA-320. (N = 3)
Cell uptake of [18F]MICA-316 and [18F]MICA-320
The ability to detect apoptosis in vitro was determined in HeLa and caspase-3-deficient MCF-7 cells. Two tracers were selected from the small library, including the least hydrophilic probe [18F]MICA-316 and the more hydrophilic tracer [18F]MICA-320. These tracers were prioritized based on the potential for optimal cellular permeability and best metabolic stability, respectively.
Treatment with STS led to a [18F]MICA-316 cell uptake in caspase-3 positive HeLa cells of 28.5 ± 5.2%ID/g, compared to 13.1 ± 7.0% in the untreated group and 10.6 ± 4.9% in the blocked group (Fig. 3A). The increase in uptake in HeLa cells was significant compared to the uptake in the treated caspase-3-deficient MCF7 cells (7.53 ± 1.53%, p = 0.0097).

A [18F]MICA-316 (black bar) and [18F]MICA-320 (grey bar) uptake in HeLa and MCF7 cells treated with STS. Results are normalised for amount of protein (N = 5). B Amount of liberated AMC from DEVD-AMC in caspase activity assay (N = 3) and activity based labelling of cell lysates with [18F]MICA-316 (N = 1)
HeLa cells stimulated with STS had a [18F]MICA-320 cell uptake of 8.5 ± 0.7%, which was significantly higher than the uptake in unstimulated cells (5.2 ± 1.3% (p = 0.0418)). Unexpectedly, pre-incubation with Ac-ATS010-KE did not lead to a significant change in tracer uptake for both [18F]MICA-316 and [18F]MICA-320. Most importantly, the cell uptake of [18F]MICA-316 was significantly higher compared to [18F]MICA-320 in STS-treated HeLa cells (p = 0.0079), and therefore, [18F]MICA-316 was used in subsequent experiments.
The validation study by caspase activity determination showed an AMC liberation in HeLa cells treated with STS of 1598 ± 444 pmol AMC compared to the controls; non-treated HeLa cells, treated HeLa cells + inhibitor, non-treated HeLa cells + inhibitor, treated MCF-7 cells, and, non-treated MCF-7 cells (33 ± 69, − 64 ± 14, − 40 ± 45, 253 ± 56, and, 95 ± 19, respectively)(Fig. 3B). Furthermore, cell lysates labelled with [18F]MICA-316 showed bands at 17 and 19 kDa for the treated HeLa cells that correspond to the caspase-3 subunits.
[18F]MICA-316 uptake in apoptotic tumours
To determine if the in vitro best performing tracer, [18F]MICA-316, was able to recognise apoptosis in vivo, the radiotracer was administered to colorectal tumour bearing mice treated with conatumumab or an IgG1-isotype control as previously described (Elvas et al. 2019). In the treated group, 24 h post treatment, a low total tumour uptake of [18F]MICA-316 was observed in the first 60 min post tracer injection (0.55 ± 0.33%ID/CC and 0.52 ± 0.12%ID/CC at 60 min p.i. for treated and IgG1-isotype control, respectively). Moreover, tumour uptake in the conatumumab-treated group was not higher than the tumour uptake measured in the baseline and the control group (Fig. 4A,C). IHC of resected tumours showed a more pronounced caspase-3 staining of the conatumumab-treated tumours than the IgG1-isotype control-treated. As observed in previous work, the conatumumab-treated tumours showed a heterogeneous caspase-3 activity with different “islands” of stained cells (Fig. 4B) (Elvas et al. 2019). Therefore, further voxel-wise analysis of the dynamic images was performed (Fig. 4D) that demonstrated a high heterogeneity of the high-intensity voxels in the treated tumours. However, no significant difference in intensity of the high-intensity voxels (P95) was observed between conatumumab-treated and IgG1 isotype control-treated tumours (p = 0.8857).

Biodistribution of [18F]MICA-316 in COLO205 tumour-bearing mice. A Representative slices of PET image 1 h p.i. of conatumumab-treated mice pre-and post-treatment and of IgG1 isotype control treated mouse pre- and post-treatment. White circles indicate tumours. B Representative 20X pictures of stained slices of COLO205 tumours treated with conatumumab (top) and IgG1 control (bottom). Slices were stained with cleaved caspase-3 antibody (brown staining). C TAC of COLO205-tumours at baseline (black), after treatment with conatumumab (blue), or, after treatment with IgG1 isotype control (red) for 60 min. Values are expressed in %ID/CC (N = 4/group). D Voxel-based analysis of tumour uptake in COLO205-tumour bearing mice. The intensities of each voxel of the tumour of the dynamic image are expressed in a box plot showing the quartiles, P5 and P95 of mice at baseline (light grey) and post-treatment (dark grey)
In contrast, SPECT imaging of [99mTc]Tc-duramycin (positive control radiotracer) showed an higher uptake of the tracer in conatumumab treated COLO205-bearing mice (1.75 ± 0.13%ID/CC) tumour model in comparison to the IgG1 isotype control treated mice (1.02 ± 0.59%ID/CC) (SI figure 32).
Activity-based labelling of tumour homogenates
Tumour homogenates were obtained from COLO205 and HT29-tumour bearing mice treated with either conatumumab or an IgG1 control for 24 h. Caspase activity was determined ex vivo on the tumour homogenates as previously described. Figure 5, showing the amount of liberated AMC for each treatment group, demonstrate that there is a higher caspase activity in the COLO205 treated tumours (15,119 ± 761 pmol liberated AMC) in comparison to the control groups (1041 ± 65 (p = 0.4249), 679 ± 63 (p = 0.0099), and, 286 ± 29 (p < 0.0001) pmol liberated AMC for COLO205 control, HT29 treated and HT29 control, respectively).

Measured amount of caspase activity in tumour homogenates using a fluorogenic substrate. Results are expressed as amount of freed AMC (pmol), (N = 3) and autoradiography image of tumour homogenates labelled with [18F]MICA-316, indicating labelling of active caspase-3 at 17 and 19 kDa for treated COLO205 tumours (N = 3)
To determine the ability of [18F]MICA-316 to label active caspase-3 in apoptotic tumours ex vivo, tumour homogenates were labelled as previously described (Elvas et al. 2019). The 17 and 19 kDa bands observed in the COLO205 treated tumour homogenate demonstrated that [18F]MICA-316 was able to bind active caspase-3 ex vivo in the tumour model. Lack of bands in the control groups suggest a negligible active caspase-3 is present in these models.
Discussion
In this study we developed novel 18F-labelled PET tracers for the imaging of caspase-3 activation for treatment response evaluation. Initially, the DW3-KE inhibitors were used as a scaffold as they were reported to have a high selectivity for caspase-3, due to the interactions of prime side KE warhead and the Phe on P2 with caspase-3. N-terminal modifications of this sequence had a limited effect on the selectivity. However, this sequence had slow binding kinetics to caspase-3 in comparison to the first generation DEVD-AOMK scaffold, which we hypothesize is the main reason for the low uptake in vivo of [18F]MICA-309. Therefore, a 3rd generation of tracers were generated ([18F]MICA-314 to -318 and -320) based on the ATS010-KE scaffold, that has a PF5 residue instead of βhLeu, on P3, resulting in a 154 × increase in kinact/Ki. The low impact of the N-terminal modifications on the reactivity of the ATS010-KE probes was expected, as a crystal structure determined that amino acids after P5’ lie outside the active site of active caspase-3, and previous N-terminal modifications such as coupling of biotin had minimal effects on the potency (Solania et al. 2019).
The radiosynthesis of [18F]MICA-314 to -318 was performed through the click reaction of [18F]Fluoro-pent-1-yne as previously described (Elvas et al. 2019). This method allows the use of a small amount of peptide precursor while still achieving adequate yields for preclinical evaluation of the tracers. Semi-prep HPLC purification resulted in a high RCP and high molar activity by separating the radiotracer from the unlabelled peptide precursor. The radiosynthesis of [18F]MICA-320 was performed with a different prosthetic group but the final step consists of CuAAC followed by semi-prep HPLC. Another advantage of these synthetic routes is that the peptides do not undergo harsh radiolabelling conditions with the potential of peptide degradation. Furthermore, the use of CuAAC results in a low amount of side-products. A disadvantage of this approach is the relatively long synthesis time (90–145 min) due to multiple steps, a 20-min click reaction, and, the semi-prep purification step followed by formulation through solid phase extraction. Improvements in the synthesis can be achieved by improving the yield of the CuAAC, using a biocompatible mobile phase, and optimizing the sequence to decrease the duration of the synthesis.
All tracers showed a favourable stability in mouse plasma in vitro over 2 h. Interestingly, a slightly lower in vitro stability of [18F]MICA-315 was detected in mouse plasma compared to the other radiotracers of the library. Instability of PEG-groups in mouse serum due to oxidation or hydrolysis has been observed by other research groups and can explain the lower stability of this radiotracer (Raheem et al. 2023). All radiotracers demonstrated a low in vivo stability with the formation of polar radiometabolites over time. Similar in vivo stability results were observed for DEVD-based radiotracers (Elvas et al. 2019; Su et al. 2013b). Although the identity of radiometabolites were not experimentally confirmed, it was hypothesized that hydrolysis of the peptide backbone by plasma hydrolases was the cause of degradation. In this study, unnatural amino acids were introduced in the peptide backbone to increase selectivity for caspase-3. It is known that the introduction of unnatural amino acids can increase the stability of peptides from proteases, which makes the lower stability of this library compared to previously developed DEVD-based radiotracers surprising (Lu et al. 2020). Nevertheless, a fast metabolization can be overcome by a fast binding of the radiotracer to the target, presumably allowing the in vivo visualization of the target. A limitation of this study is that the radiometabolites in the excretions were not determined providing us no information on the difference in excretion of the intact tracer compared to the radiometabolites.
The PK profiles of all ATS010-KE-derived radiotracers were similar in healthy CD1−/− nude mice with fast clearance from the blood and mixed renal-hepatobiliary excretion, similarly to the PK profile of the DEVD-based ABP [18F]MICA-302 (Elvas et al. 2019). It had been hypothesized before that the high intestinal activity could be due to caspase activity in the small intestines. However, the PET images indicate that the activity in the small intestines is mostly due to hepatobiliary excretion instead of caspase binding, as the activity is seen in different parts of the small intestines over time. Furthermore, the faeces of 5 mice injected with [18F]MICA-316 was collected and showed a high amount of radioactivity (data not shown). Fast blood clearance further validates the low plasma protein binding observed in the stability study, which is beneficial for increasing target-to-background ratio. However, a low blood retention time limits the time the tracer can accumulate to the tumour.
In the in vitro cell uptake experiment, the uptake of [18F]MICA-316 and [18F]MICA-320 in HeLa cells treated with STS could not be significantly blocked by pre-incubation with the Ac-ATS010-KE inhibitor. A hypothesis is that there is important unspecific binding of the tracers to other cell components. Nevertheless, it is interesting that such a large difference between [18F]MICA-316 and [18F]MICA-320 uptake in STS-treated HeLa cells was observed in this in vitro assay, although both radiotracers are slightly hydrophilic (− 0.42 ± 0.18 and − 1.20 ± 0.19 for [18F]MICA-316 and [18F]MICA-320, respectively). It is important for the development of the next generation of caspase-3-selective ABPs to consider the effect of novel moieties on the lipophilicity, and therefore, the cell uptake.
Fast binding kinetics in vitro and a high selectivity over other caspases, the ability to bind caspase-3 in cell and tumour homogenates, and, the increased uptake in apoptotic cells compared to control cells, motivated us to test [18F]MICA-316 in vivo in a tumour mouse model. However, the radiotracer was unable to discriminate between conatumumab-treated and IgG1 isotype control-treated mice. Furthermore, IHC and voxel-based analysis demonstrated that the caspase-3 activation and the radioactivity distribution in the tumours were heterogenous. Nevertheless, no difference between the high activity voxels of both groups was observed. We hypothesize that the main reason for the lack of discrimination between the conatumumab and IgG1 isotype control-treated is due to low total uptake of radiotracer in the tumour. Another possibility is that the low in vivo stability results in a low amount of intact tracer to bind the intracellular activated caspase-3. Nevertheless, positive imaging results have been observed with less stable radiotracers (Adhikari et al. 2023). A final hypothesis, is that due to caspase-3 activation being a transient process, and, the blebbing of the cell membrane during late apoptosis leading to efflux of protein, that caspase-3 is not an ideal target for PET imaging. Nevertheless, other groups have reported an increased tracer uptake in vivo in tumour cell death models by targeting caspase-3, disproving this latter hypothesis (Chen et al. 2021; Lin et al. 2022a). A lower uptake of the cell death tracer [99mTc]Tc-duramycin (positive control) was observed in the current study (about 3 × lower) compared to our previous studies, as well as a less pronounced cc-3 colouring of the tumour tissue, indicating that the treatment was less effective than in our previous studies, supporting the idea that lack of target availability could be the reason for low tumour uptake (Elvas et al. 2017).
Nonetheless, the PE-binding radiotracer, [99mTc]Tc-duramycin, was able to discriminate the conatumumab-treated from the IgG1 isotype control treated mice by binding PE from the dying cells, making it the best treatment response evaluating probe thus far. Due to the higher spatial resolution of PET tracers in comparison to SPECT tracers, there is still room for improvement. To this purpose, different groups have already successfully attempted to develop 18F- and 68 Ga-labelled duramycin (Wiele et al. 2021). Nevertheless, caspase-3 remains an interesting target for molecular imaging, as it is the main effector of apoptosis, but also has a role in different processes (Eskandari and Eaves 2022).
Further optimisation of our caspase-3 tracer library will be performed to increase metabolic stability or increase tumour uptake. Metabolic stability can be increased by introducing D-amino acids in the backbone or creating the full sequence with D-amino acids through retro-inversion, however, a loss of target affinity is possible and binding kinetics have to be thoroughly tested. Another strategy that can be followed is the methylation of amides, further decreasing protease recognition. However, due to the hydrogen bonds formed between the ABP and caspase-3, realistically only methylation of the amide between P2 and P3 or after P5 could improve metabolic stability without loss of target affinity (Solania et al. 2019). By substituting some amides for more stable sulfonamides, a decreased metabolization of the ABPs while retaining hydrogen bond donor characteristics can be achieved. Finally, increasing steric hindrance after P5 could result in an increased stability against proteases while not dramatically influencing binding kinetics. One can imagine that any modifications of the backbone will not only impact target affinity, but also the biodistribution and cell uptake of the tracers (Evans et al. 2020). In addition to increasing metabolic stability, increasing the initial uptake in the tumour environment is a strategy that can lead to an increased ABP concentration in the tumour and an increased tracer retention, leading to a higher tumour-to-background ratio. An increase in initial tumour uptake can be achieved by using a targeting moiety (Su et al. 2021).
Conclusion
In conclusion, a library of novel 18F-labelled, caspase-3 selective ABPs was developed for treatment response evaluation based on the Ac-DW3-KE and Ac-ATS010-KE inhibitors, retaining the kinetic properties of the original inhibitors. [18F]MICA-316, was able to detect apoptosis in an in vitro cell death assay, however, low tumour accumulation was observed in vivo making this tracer unsuitable for clinical translation.
Availability of data and material
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ABP:
-
Activity-based probe
- Ac:
-
Acetyl
- Acp:
-
6-Amino-hexanoic acid
- Am:
-
Molar activity
- AMAC:
-
2-Aminoacridin-9(10H)-one
- AMC:
-
Amino-4-methylcoumarin
- AUC:
-
Area under the curve
- BCA:
-
Bichinconic acid
- CT:
-
Computed tomography
- CuAAC:
-
Copper-catalysed azide-alkyne cycloaddition
- E:
-
Glutamic acid
- EOB:
-
End of bombardment
- G:
-
Glycine
- HPLC:
-
High-performance liquid chromatography
- HRMS:
-
High resolution mass spectrometry
- IHC:
-
Immunohistochemistry
- irRC:
-
Immune-related response criteria
- kobs :
-
Rate constant for the conversion from the initial velocity phase to the steady state velocity phase
- Ki :
-
Concentration of inhibitor that yields ½ kinact
- kinact :
-
Maximum rate of inactivation
- MRI:
-
Magnetic resonance imaging
- NMR:
-
Nuclear magnetic resonance
- PE:
-
Phosphatidylethanolamine
- PEG:
-
Polyethylene glycol
- PET:
-
Positron emission tomography
- PK:
-
Pharmacokinetics
- PS:
-
Phosphatidylserine
- p.i.:
-
Post-injection
- RECIST:
-
Response Evaluation Criteria in Solid Tumours
- RCP:
-
Radiochemical purity
- RCY:
-
Radiochemical yield
- SBP:
-
Substrate-based probe
- SPECT:
-
Single photon emission computed tomography
- STS:
-
Staurosporine
- TAC:
-
Time-activity curve
- V0 :
-
Initial velocity
- Vs :
-
Steady-state velocity
- VOIs:
-
Volumes of interest
- %ID/CC:
-
Percent inject dose per cubic centimeter
References
Adhikari K, Dewulf J, Vangestel C, et al. Characterization of structurally diverse 18F-labeled d-TCO derivatives as a PET probe for bioorthogonal pretargeted imaging. ACS Omega. 2023;8:38252–62.
Beroske L, Van den Wyngaert T, Stroobants S, Van der Veken P, Elvas F. Molecular imaging of apoptosis: the case of caspase-3 radiotracers. Int J Mol Sci. 2021;22:22.
Carter BW, Bhosale PR, Yang WT. Immunotherapy and the role of imaging. Cancer. 2018;124:2906–22.
Chen DL, Zhou D, Chu W, et al. Comparison of radiolabeled isatin analogs for imaging apoptosis with positron emission tomography. Nucl Med Biol. 2009;36:651–8.
Chen DL, Engle JT, Griffin EA, et al. Imaging caspase-3 activation as a marker of apoptosis-targeted treatment response in cancer. Mol Imaging Biol. 2015;17:384–93.
Chen Z, Chen M, Zhou K, Rao J. Pre-targeted imaging of protease activity through in situ assembly of nanoparticles. Angew Chem Int Ed. 2020;59:7864–70.
Chen M, Chen Z, Castillo JB, et al. [18 F]-C-SNAT4: an improved caspase-3-sensitive nanoaggregation PET tracer for imaging of tumor responses to chemo-and immunotherapies. Eur J Nucl Med Mol Imaging. 2021;48:3386–99.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 11). Eur J Cancer. 2009;45:228–47.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516.
Elvas F, Vangestel C, Pak K, et al. Early prediction of tumor response to treatment: preclinical validation of 99mTc-Duramycin. J Nucl Med. 2016;57:805–11.
Elvas F, Boddaert J, Vangestel C, et al. 99mTc-Duramycin SPECT imaging of early tumor response to targeted therapy: a comparison with 18F-FDG PET. J Nucl Med. 2017;58:665–70.
Elvas F, Vanden Berghe T, Adriaenssens Y, et al. Caspase-3 probes for PET imaging of apoptotic tumor response to anticancer therapy. Org Biomol Chem. 2019;17:4801–24.
Engel BJ, Gammon ST, Chaudhari R, et al. Caspase-3 substrates for noninvasive pharmacodynamic imaging of apoptosis by PET/CT. Bioconjug Chem. 2018;29(9):3180–95.
Eskandari E, Eaves CJ. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J Cell Biol. 2022;221: e202201159.
Evans BJ, King AT, Katsifis A, Matesic L, Jamie JF. Methods to enhance the metabolic stability of peptide-based PET radiopharmaceuticals. Molecules. 2020;25:2314.
Glaser M, Rajkumar V, Diocou S, et al. Biological evaluation of a caspase-3 selective 5-[123,125 I]iodo-1,2,3-triazole derived isatin Spect tracer. Sci Rep. 2019;9:19299.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hight MR, Cheung YY, Nickels ML, et al. A Peptide-based positron emission tomography probe for in vivo detection of caspase activity in apoptotic cells. Clin Cancer Res. 2014;20:2126.
Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020;6:112.
Kummar S, Gutierrez M, Doroshow JH, Murgo AJ. Drug development in oncology: classical cytotoxics and molecularly targeted agents. Br J Clin Pharmacol. 2006;62:15–26.
Lin J, Gao D, Wang S, et al. Stimuli-responsive macrocyclization scaffold allows in situ self-assembly of radioactive tracers for positron emission tomography imaging of enzyme activity. J Am Chem Soc. 2022a;144:7667–75.
Lin J, Gao D, Wang S, et al. Stimuli-responsive macrocyclization scaffold allows in situ self-assembly of radioactive tracers for positron emission tomography imaging of enzyme activity. J Am Chem Soc. 2022b;144:7667–75.
Lu J, Xu H, Xia J, et al. D- and unnatural amino acid substituted antimicrobial peptides with improved proteolytic resistance and their proteolytic degradation characteristics. Front Microbiol. 2020;11: 563030.
Miranda A, Bertoglio D, Glorie D, Stroobants S, Staelens S, Verhaeghe J. Validation of a spatially variant resolution model for small animal brain PET studies. Biomed Phys Eng Express. 2020;6: 045001.
Mohammad RM, Muqbil I, Lowe L, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35:S78.
Morana O, Wood W, Gregory CD. The Apoptosis paradox in cancer. Int J Mol Sci. 2022;23:1328.
Nguyen QD, Smith G, Glaser M, Perumal M, Årstad E, Aboagye EO. Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-3/7 specific [18F]-labeled isatin sulfonamide. Proc Natl Acad Sci USA. 2009;106:16375–80.
Ostapchenko VG, Snir J, Suchy M, et al. Detection of active caspase-3 in mouse models of stroke and Alzheimer’s disease with a novel dual positron emission tomography/fluorescent tracer [68 Ga]Ga-TC3-OGDOTA. Contrast Media Mol Imaging. 2019;2019:6403274.
Qiu L, Wang W, Li K, et al. Rational design of caspase-responsive smart molecular probe for positron emission tomography imaging of drug-induced apoptosis. Theranostics. 2019;9:6962.
Raheem SJ, Salih AK, Dominguez Garcia M, Sharpe JC, Toosi BM, Price EW. A systematic investigation into the influence of net charge on the biological distribution of radiometalated peptides using [68Ga]Ga-DOTA-TATE Derivatives. Bioconjugate Chem. 2023;34:561.
Rapic S, Vangestel C, Elvas F, et al. Evaluation of [18F]CP18 as a substrate-based apoptosis imaging agent for the assessment of early treatment response in oncology. Mol Imaging Biol. 2017;19:560–9.
Shen B, Jeon J, Palner M, et al. Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-triggered nano-aggregation probe. Angew Chem Int Ed Engl. 2013;52:10511.
Smith G, Glaser M, Perumal M, et al. Design, synthesis, and biological characterization of a caspase 3/7 selective isatin labeled with 2-[18f]fluoroethylazide. J Med Chem. 2008;51:8057–67.
Solania A, González-Paéz GE, Wolan DW. Selective and rapid cell-permeable inhibitor of human caspase-3. ACS Chem Biol. 2019;14:2463–70.
Stéen EJL, Jørgensen JT, Denk C, et al. Lipophilicity and click reactivity determine the performance of bioorthogonal tetrazine tools in pretargeted in vivo chemistry. ACS Pharmacol Transl Sci. 2021;4:824–33.
Su H, Chen G, Gangadharmath U, et al. Evaluation of [18 F]-CP18 as a PET imaging tracer for apoptosis. Mol Imaging Biol. 2013a;15:739–47.
Su H, Chen G, Gangadharmath U, et al. Evaluation of [18F]-CP18 as a PET imaging tracer for apoptosis. Mol Imaging Biol. 2013b;15:739–47.
Su Z, Xiao D, Xie F, et al. Antibody-drug conjugates: recent advances in linker chemistry. Acta Pharm Sin B. 2021;11:3889–901.
Udemba A, Smith G, Nguyen Q-D, et al. Organic & Biomolecular Chemistry Design, synthesis and initial characterisation of a radiolabelled [18 F]pyrimidoindolone probe for detecting activated caspase-3/7 †. Org Biomol Chem. 2015;13:5418.
Van de Wiele C, Ustmert S, De Spiegeleer B, De Jonghe PJ, Sathekge M, Alex M. Apoptosis imaging in oncology by means of positron emission tomography: a review. Int J Mol Sci. 2021;22:1–14.
Vickers CJ, Gonzaíez-Paéz GE, Wolan DW. Selective detection and inhibition of active caspase-3 in cells with optimized peptides. J Am Chem Soc. 2013;135:12869–76.
Vickers CJ, González-Páez GE, Wolan DW. Discovery of a highly selective caspase-3 substrate for imaging live cells. ACS Chem Biol. 2014;9:2199–203.
Waldmann CM, Hermann S, Faust A, et al. Novel fluorine-18 labeled 5-(1-pyrrolidinylsulfonyl)-7-azaisatin derivatives as potential PET tracers for in vivo imaging of activated caspases in apoptosis. Bioorg Med Chem. 2015;23:5734–9.
Zhou D, Chu W, Rothfuss J, et al. Synthesis, radiolabeling, and in vivo evaluation of an 18F-labeled isatin analog for imaging caspase-3 activation in apoptosis. Bioorg Med Chem Lett. 2006;16:5041–6.
Acknowledgements
The authors gratefully acknowledge Ingrid De Meester and Emile Verhulst with the technical support for the kinetic measurement of 9, and, Philippe Joye, Caroline Berghmans, Eleni Van der Hallen, and Annemie Van Eetveldt for their assistance with small animal handling, PET scans and IHC.
Funding
This work was funded by the Bijzonder onderzoeksfonds (BOF), University of Antwerp (DOCPRO 46060) and the Research Foundation Flanders (FWO) (postdoctoral fellowship 12T8821N).
Author information
Authors and Affiliations
Contributions
For this manuscript, the 12 authors contributed profoundly. FE, LW and DW conceived and designed the study. LL, LB, KA and FE synthesized the compounds. LL, LB, NV, AM and AS acquired the data. LL, LB, NV, AS and FE analysed and interpreted the data. PVdV and FE supervised the study. AL contributed to the kinetic measurements. LL wrote the manuscript. FE acquired funding. CV and FE critically edited and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Experimental procedures and protocols were performed following European Directive 86/609/EEC Welfare and Treatment of Animals and were approved by the local ethical committee (2018-45, University of Antwerp, Belgium).
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lauwerys, L., Beroske, L., Solania, A. et al. Development of caspase-3-selective activity-based probes for PET imaging of apoptosis. EJNMMI radiopharm. chem. 9, 58 (2024). https://doi.org/10.1186/s41181-024-00291-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41181-024-00291-x