
Abstract
An understanding of the mechanisms of Ti is incorporation into silicate glasses and melts is critical for the field of petrology. Trace-element thermobarometry, high-field-strength element partitioning, and the physical properties of magmas are all be influenced by Ti incorporation into glasses and changes therein in response to changes in composition and temperature. In this study, we combine 29Si solid state NMR and Ti K-edge XAFS spectroscopy to investigate how Ti is incorporated into quenched Na-silicate glasses, and the influence of Ti on the structure of silicate species in these glasses. 29Si NMR shows that in both Ti-bearing Na2O•4SiO2 (NS4) and Na2O•8SiO2 (NS8) glasses, increasing the amount of Ti in the melt results in a shift of Si Q4 peak in the 29Si NMR spectra reflecting Ti nearest neighbors for Si in Q4 speciation. The Ti XAFS results from NS8 glass indicate that Ti is primarily incorporated in [5]-fold coordination. At higher Ti content, there is a shift of the XAFS pre-edge feature suggesting mixing of [4]-fold Ti into the spectra. Combined, the 29Si NMR and XAFS pre-edge data are consistent with Ti incorporation as isolated [5]Ti atoms and the formation of [5]Ti clusters at relatively low Ti concentrations, with no evidence for Ti–Na interactions as suggested by previous studies. As the Ti content increases, the Ti atoms begin to occupy 4-fold coordinated sites interacting primarily with Si in Q4 speciation (no significant Na–[4] Ti bonding). The internal consistency of these two techniques provides a uniquely complete snapshot of the complexity of Ti incorporation in silicate melts and underlies the importance of understanding Ti incorporation mechanisms in natural magmatic systems.

Similar content being viewed by others

Introduction
Titanium, typically a minor element in natural magmatic systems, does nevertheless, play a major role in the evolution of igneous and metamorphic rocks. Ti-rich oxide minerals control trace-element budgets in magmas and metamorphic melting reactions (Ryerson and Watson 1987; Tang et al. 2019; Xiong 2006), Ti activity in magmas is a critical component of trace-element thermobarometry (Ackerson et al. 2018; Ghiorso and Gualda 2013; Watson and Harrison 2005), and Ti incorporation imparts unique structural and physical properties (e.g., refractive index, tensile strength, compressibility) into melts and glasses (Liska et al. 1996; Liu and Lange 2001; Morsi and El-Shennawi 1984; Roskosz et al. 2004; Scannell et al. 2015). All these characteristics are controlled by the mechanisms of Ti incorporation (solubility) into melts/glasses, yet our understanding of these solubility mechanisms is fundamentally limited.
The Ti solubility in magmatic liquids varies over many orders of magnitude, from 1000 s of ppm in silicic magmas (Hayden and Manning 2011; Hayden and Watson 2007) to over 20 wt% in intermediate and mafic glasses (Gaetani et al. 2008). This behavior is likely due to the number of potential solubility mechanisms for Ti in magmas. In various natural and synthetic glasses and using multiple analytical techniques [e.g., Raman (Henderson and Fleet 1995; Mysen and Richet 2005), XAFS (Farges 1997; Farges et al. 1996a), molar volume calculations (Lange and Navrotsky 1993)], Ti has been demonstrated to be incorporated in [4]-, [5]-, and [6]-fold coordination. In [4]-fold coordination Ti acts primarily as a network-forming cation, helping to polymerize the melt, whereas [6]-fold coordinated Ti will act as a network-modifying cation (Mysen and Richet 2005, references therein). Ti also has the potential to cluster in melts, particularly in [5]- and [6]-fold coordination, where [5]Ti is suggested to form Na-titanite isolates and clusters (Farges et al. 1996a; Yarker et al. 1986).
One potential way to investigate Ti solubility and its influence on melt structure is to observe the effect of Ti on other cations in quenched glasses. 29Si NMR provides the opportunity to do this, as it can be used to resolve how addition of Ti to magma will affect the degree of polymerization and the bonding environment of Si. A combination of 29Si NMR and Ti K-edge XAFS pre-edge spectroscopy (which provides information on the average coordination of Ti in glasses) can give a more complete picture of how Ti is incorporated into silicate glasses. In this study, we utilized these two techniques on a series of synthetic Ti-bearing Na-silicate glasses to observe how Ti is incorporated into glasses formed by temperature quenching of melt and how Ti solubility mechanisms may be affected by Ti concentration.
Experimental and methods
Titano-sodium silicate synthesis
In this study we have focused on investigating the perturbation in silicate melt structure (as observed in quenched glasses) through the addition of TiO2 along a simple composition join linking a binary sodium silicate composition with TiO2 (Fig 1). We focus on two specific compositions, those of Na2O•8SiO2 (NS8) and Na2O•4SiO2 (NS4), with increasing mol% addition of TiO2 from 2.5 up to 15 mol% (Fig. 1). Note that at concentrations above 15 mol%, we are close to the liquid-liquid miscibility gap (Glasser and Maar 1979). Glasses were synthesized from SiO2, TiO2, and Na2CO3 starting material by heating at a rate of 100 °C/h from 700–900 °C, then at a rate of 150 °C/h to 1400 °C in Pt crucibles. Melts were held at 1400 °C for 1 h, after which they were quenched to room temperature. In air, the glasses quenched to below the glass transition temperature in less than 30 s, owing to the small size of the Pt crucibles used. The glass structure recorded in the glasses is that of the glass at the glass transition temperature. For this study, and because of the potential for temperature dependence on the solubility mechanisms (Lange and Navrotsky 1993), all glasses were synthesized at 1400 °C for 1 h. Several NS8 glasses with 10 mol% TiO2 were also synthesized at multiple temperatures to test for a potential temperature dependence on XAFS spectra (supplementary fig. S2). The glasses were inspected in immersion oil under a petrographic microscope to ensure no crystalline phases were present, and to ensure there were no signs (e.g., cloudiness, opalescence) indicating the presence of nanocrystalline anatase (Henderson and Fleet 1995). Furthermore, no anatase was detected in these glasses in X-ray diffraction (XRD) spectra (Ackerson and Mysen in press).

Upper portion of the SiO2–Na2O–TiO2 phase diagram. Our experiments were performed along the NS8 and NS4 joins. Experimental bulk compositions are denoted with black circles. Thin black lines are liquidus isotherms, and thick black lines are liquidus phase boundaries. (modified from Glasser and Maar 1979)
29Si solid state nuclear magnetic resonance spectroscopy
All solid state 29Si NMR spectra were acquired using a Chemagnetics Infinity solid state NMR spectrometer that employs a 7 Tesla static magnetic field. The resonant frequency of 29Si at ~ 7 Tesla is 59.6 MHz. Crushed glass samples were loaded into a 5 mm diameter rotor and placed within a Chemagnetics double resonance magic angle spinning (MAS) probe. Following Maekawa et al. 1991, the addition of a paramagnetic relaxation agent (Fe2O3 at 0.1 wt%) results in a significant reduction in 29Si’s spin lattice relaxation (T1) time without spectral distortion. Tests on NS4 glass revealed that a pulse width of 1.3 μs, corresponding to a 30° nutation angle and recycle delays spanning from 2 to 20 s did not reveal any reduction in intensity resulting T1 saturation; hence, a recycle delay of 2 s was chosen for all compositions. The MAS frequency (ωr/2π) was 8 KHz. The number of acquisitions was in each case 40 K and the 29Si spectra were referenced to the 29Si frequency of tetramethylsilane (TMS) defined as 0 ppm.
Ti K-edge pre-edge X-ray absorption fine structure (XAFS)
XAFS spectra were collected on a suite of Ti-bearing NS8 glasses at beamline 13 IDE at the Advanced Photon Source at Argonne National Laboratory. The beam was filtered using the Si (111) monochromator, tuned to the absorption edge at 4964.46 eV, and was calibrated on Ti foil. Relative to the absorption edge, scans were conducted over a dynamic range with an emphasis on collecting high energy resolution spectra in the pre-edge region: from − 60 to − 6 eV data were collected at a 2 eV step for 2 s, from − 6 to 15 eV at a 0.1 eV step for 2 s, and a 2 eV step for 2 s from 15 to 200 eV. Data were processed using the Larch software (Newville 2013). Each spectrum was processed by first defining the absorption edge at the maximum first derivative of the spectra within the edge step region (care was taken not to inadvertently select the maximum first derivative of the pre-edge feature), then applying a linear normalization to the pre-edge region (set to 0) and edge step (set to 1), focusing on normalization of the XAFS region (see Ackerson et al. 2017 for normalization details).
Pre-edge peaks were fit with pseudo-Voigt peaks to determine the precise position of the peak centers (Farges et al. 2004). Linear mixing was performed for three end-member pre-edge spectra to determine the position of the pre-edge peak as a function of mixing coordination states. End-member Ti-bearing forsterite ([4]-fold), fresnoite ([5]-fold), and titanite ([6]-fold) were used as end-members for mixing curves. Peak heights of the primary peak were used to determine mixed peak positions in lieu of peak centroids since [6]-fold coordinated Ti end-member spectra have multiple discrete pre-edge peaks. Importantly, the influence of these complex features on spectra diminishes rapidly during mixing, so their net influence on the spectra is less than the precision of the mixing technique (i.e., the ~+/− 10% mixing estimates for coordination mixing is greater than the influence of complex [6]-fold peaks).
Results and discussion
The NS4 and NS8 compositions utilized in this study have been extensively studied for the purpose of elucidating the influence of volatiles and network-forming and network-modifying cations on melt structure (Cody et al. 2005; Kuemmerlen et al. 1992; Maekawa et al. 1991; Mysen et al. 2011; Roskosz et al. 2006; Zotov and Keppler 1998), and were chosen specifically as they provide a model system for studying moderately polymerized and chemically evolved magmatic liquids. Additionally, sodium silicate compositions are favored for structure vs. composition studies is because the key structural elements of silicate glasses and melts (the so called “Qn” species (Lippmaa et al. 2002), see discussion below) are clearly resolved using 29Si solid state NMR.
Sodium is a network modifying cation, which means that addition of Na2O to a silicate structure will yield non-bridging oxygens (NBOs). The structure of silicate melts and glasses quenched from them is characterized, in part, by the number of bridging oxygens per silicate oxide tetrahedron. These are often described as Qn-species, which are defined by the number of bridging oxygens per tetrahedrally coordinated cations of the silicate structure (n = no. of bridging oxygens, e.g., Q4-Q0). In the case of the sodium silicate compositions explored here, given the relatively high silica content, the 29Si solid state NMR reveals only Q3 and Q4 species well resolved from each other (Cody et al. 2005; Kuemmerlen et al. 1992; Maekawa et al. 1991; Zotov and Keppler 1998).
Silicon (29Si) solid state NMR data
29Si is the only silicon isotope (of three: 28Si, 29Si, and 30Si) with spin (S = 1/2 in this case), with a natural abundance of 4.7%. In the present case, all silicon is in the 4+ valence state and tetrahedrally bonded to four oxygen atoms.
In simple silicate systems such as NS8-TiO2 and NS4-TiO2, there are three primary factors that affect the 29Si solid state NMR spectrum. First, there is the approximately + 10 ppm shift accompanying changes in NBO (i.e., moving from Q4 to Q3 to Q2 … etc.) (Maekawa et al. 1991; Magi et al. 1984) (Fig. 2). As noted by Maekawa et al. 1991, there is a systematic shift in Q4 and Q3 frequency with increasing sodium content, where the range in shift is greatest for Q4 and decreases with increased NBOs. For any given NS composition, the variation in shift (evident in bandwidth) is much less than indicated in Fig. 2: whereas the bands at the top of the figure show bandwidth for a wide range of NS compositions, the positions of Q4 and Q3 for NS8 and NS4 much narrower (shown with gray regions in Fig. 2).

Factors influencing peak positions of Q-species in 29Si NMR spectra. Grey vertical bars are the peak frequencies observed in the present study. The number of non-bridging oxygens (number of Si–O–Na interactions) causes a positive shift in peak frequency, such that Si in Q2 speciation (2 non-bridging oxygens) is at a higher frequency than Si in Q4 speciation (0 non-bridging oxygens). For Si in Q4 speciation, Al as a network-forming cation nearest neighbor has been shown to cause a shift toward higher frequency. Q4 Si with one Al nearest neighbor (Al1) is at a lower frequency than Q4 Si with 4 Al nearest neighbors (Al4). Variations on Si–O–Si bond angle can also induce large shifts in the Q4 peak position, shown here from lower frequency (− 122 ppm at 170°) to higher frequency (− 93 ppm at 130°). These variations in bond angle are the cause of the Al nearest neighbor effects (Lippmaa et al. 1981)
A second factor that governs 29Si shift (in the present case) is the perturbation to the 29Si frequency related to substitution of next nearest network cations with elements other than silicon (e.g., with aluminum) as has been studied by Lippmaa et al. 1981 (Fig. 2). In this case, the effect on 29Si frequency in a Q4 species with sequential substitution Al3+ for Si4+ induces a ~+ 5 ppm shift per substitution (Fig. 2) (Lippmaa et al. 1981). The physics underlying this shift is understood to involve differences in the electronegativity of Al3+ relative to Si4+ (Janes and Oldfield 1985). While this effect is well documented for aluminum substitution, as far as we can find, no such information is available for the perturbation resulting from titanium (Ti4+) substitution for Si4+. We do note, however, that the electronegativity of Ti4+ is much closer to that of Al3+ (Pauling 2014) than Si4+, we, therefore, expect the perturbation on the 29Si NMR signal due to titanium nearest neighbors to be similar to that of aluminum substitution.
The third major perturbation on 29Si NMR frequencies is any variation in Si–O–Si bond angle. As shown in Fig. 2 for 29Si in Q4 a variation in Si–O–Si angle from 170 to 130° results in a range in frequency from − 122 ppm up to a frequency of − 93 (Mauri et al. 2000; Smith and Blackwell 1983). This is just for one Q species. If multiple Q species are present with such variation, the 29Si NMR spectrum becomes extremely broad and detailed structural interpretation is very difficult. It is notable that wide variation in NS Q4 (Fig. 2) 29Si NMR frequency as well as that observed for Si Q4 (Al0) is most likely due to variation in Si–O–Si bond angle. For example, (Le Losq et al. 2015) showed that lithium, sodium, and potassium tetrasilicate glasses, there is a systematic shift in Si–O–Si bond angles in Q4 species, where from Li+ to K+ the bond angles decreased.
Figures 3 and 4 present 29Si solid state NMR spectra of the TiO2 containing NS glasses (NS8 and NS4). In the case of the NS4 glass (Fig. 3), one observes (at 0% TiO2), two peaks corresponding to Q4 and Q3 species, at − 105.5 and − 93.3 ppm, respectively. Note that although the Q3 peak appears more pronounced than the Q4 peak, the Q4 peak is slightly broader and the peak areas for NS4 are nearly identical as would be predicted based on stoichiometry. With increasing TiO2, there is an immediate and obvious shift of the Q4 peak to slightly higher frequency, while no such apparent shift in the Q3 peak is evident. In the case of the NS8 glass (Fig. 4), with increasing TiO2, there is also shift in the Q4 peak to slightly higher frequency, but the magnitude of the shift with increasing TiO2 is less than that of the NS4 + TiO2 glasses. As with the NS4 glasses, there is no obvious change in frequency or relative intensity of the Q3 species in NS8 + TiO2 glasses. The shift of the Q4 peak position in NS4 and NS8 glasses to progressively higher frequencies with increasing TiO2 is shown in Fig. 5.

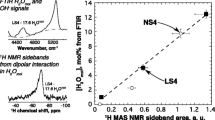
Stacked 29Si NMR data for NS4 glass at various Ti concentrations, normalized by integrated intensity. The two predominant peaks are the Q3 and Q4 peaks at − 93 and − 106 ppm, respectively. At 0% TiO2, the two peaks account for nearly equal areas under the curve, as is expected for NS4 glasses with 50% Q3 Si and 50% Q4 Si. There is a shift in peak position of the Q4 to higher frequency with addition of Ti to the NS4 glass, whereas the Q3 peak is negligibly affected by the addition of Ti. The shift in Q4 to higher frequency is likely the result of Ti nearest neighbor effects on the Q4 Si. Open circles are fitted peak centers

Stacked 29Si NMR data for NS8 glass at various Ti concentrations, normalized by integrated intensity. Peaks at − 96 and − 109 ppm are Q3 and Q4, respectively. Compared with the NS4 glasses, Ti-free NS8 glass exhibits a more pronounced Q4 peak, as is predicted based on the Na:Si of the glass. As with the NS4 glasses, there is a progressive positive shift in peak energy of the Si Q4 peak, likely as a result of Ti nearest neighbors for the Si Q4 species. Open circles are fitted peak centers

Position of the 29Si NMR Q4 peak as a function of bulk TiO2 content of the glasses for NS4 (blue) and NS8 (black) compositions. The systematic shift in frequency is likely due to the continuous growth of an additional Q4 peak due to [4]Ti nearest-neighbor interactions with Q4 Si. Peak fitting of the spectra was performed using Gaussian peaks, with fit iterations converging at a chi-squared tolerance value of 1E-6
Ti XAFS Spectroscopy
Ti K-edge XAFS pre-edge spectra can provide information on the average coordination of Ti in minerals and glasses. The pre-ionization edge feature of the Ti K-edge XAFS spectra is produced by 1 s-3d orbital transitions, where the intensity of the peak is positively correlated with the degree of d-p orbital mixing (which will be greater with decreasing coordination), and the peak energy is negatively correlated with d-p mixing (Farges et al. 1996b; Waychunas 1987). The peak intensity and energy of the pre-edge peaks for the three coordination states for Ti in glasses ([4]-, [5]-, and [6]-fold coordination) are, therefore, resolvable, making these energies useful for estimating the average Ti coordination in the quenched glasses considered here.
In previous studies of Ti-bearing glasses, pre-edge features of Ti-bearing glasses have shown that Ti exists in multiple coordination states ranging from nearly complete octahedral to complete tetrahedral coordination (Farges et al. 1996b; Romano et al. 2000). Mixed Ti coordination results in pre-edge features that are linear combination of the end-member spectra, a phenomena which can be utilized to estimate the amount of Ti in multiple coordination states (Farges 1997). In the present study, we employed XAFS to analyze selected glasses from the NS8 series and several mineral standards of known Ti coordination to verify the nature of Ti coordination and explore the potential for coordination mixing in the glasses as a function of bulk TiO2 content (Fig. 6).

Ti k-edge X-ray absorption fine structure (XAFS) pre-edge peak positions and mixing curves. The Ti pre-edge feature is sensitive to the coordination environment and is used to assess average Ti4+ coordination in the experimental glasses. Grey regions and grey, filled symbols are the pre-edge peak positions of minerals and glasses with known Ti coordination (Farges et al. 1996b). White filled symbols are mixing curves between end-member [4]Ti-bearing forsterite, [5]Ti-bearing fresnoite, and [6]Ti-bearing titanite. Red symbols are the peak positions of NS8 glasses with the mol% TiO2 indicated next to the symbols. As Ti concentration increases, the peak positions shift along a trend similar to the mixing of [4]Ti and [5]Ti, indicating the average coordination of Ti in the glasses decreases with increasing bulk TiO2 content
For a given end-member coordination state, there is significant variability in the pre-edge peak intensity and energy, which limits the precision of determining the degree of coordination mixing (Farges 1997; Farges et al. 1996b) for systems like the NS glasses studied here whose end-member Ti coordination peak positions and energies are unknown. In lieu of known end-member composition for Na-silicate glasses for any coordination state, we utilize forsterite, fresnoite, and titanite as Ti4+ [4]-, [5]-, and [6]-fold coordination standards, respectively.
For the NS8 glasses analyzed, there is a subtle but significant trend in the pre-edge peak shifting toward lower energy and slightly lower intensity with an increase in bulk TiO2. At low Ti content, the peak position is closest to end-member fresnoite, indicating that almost all Ti occurs in [5]-fold coordination. As Ti content increases, the peak systematically shifts toward lower energy by ~ 0.35 eV. As seen in the end-member mixing curves (Fig. 6), this trend is similar (albeit shifted) to the trend in mixing ~ 40% [4]Ti to end-member [5]Ti, which from the known compound model mixing will produce a ~ 0.33 eV shift. There is no indication from these results that significant [6] Ti exists in the NS8 glasses. XANES spectra also confirm that the material is glassy, with no micro- or nano-scale crystallization (supplementary figure S1).
Results from Ti pre-edge XAFS and 29Si NMR provide three critical observations that aid in interpreting structural changes in the glasses with changes in bulk TiO2 content:
- 1)
At low TiO2 content, Ti is primarily in [5]-fold coordination. With increasing bulk TiO2, the amount of [4]-fold coordinated Ti increases;
- 2)
With increasing Ti, there is no apparent growth of the 29Si Q4 peak, but rather an apparent shift toward higher energy of the Q4 peak;
- 3)
There is no frequency shift, peak growth, or apparent peak diminution of the 29Si Q3 with variations in TiO2 content.
Previous research into Ti-bearing NS2 and NS4 glasses suggests that [5]Ti is incorporated as Na-titanite complexes (Farges et al. 1996a). Na-titanite formation in NS4 and NS8 glasses would consume Na+ and result in an observed polymerization of Si via the growth of the 29Si Q4 peak at the expense of Q3 Si in the NMR spectra, which is not observed in either the NS4 or NS8 glasses. In a Ti-free NS8 glass, 25% of Si atoms are bonded to non-bridging oxygens through Si–O–Na interactions. Removal of Na+ from these bonds via Na-titanite complexes would force the polymerization of Si. In the Si29 NMR spectra, this would be seen in the growth of the Q4 peak at the expense of the Q3 peak. For example, if each [5]Ti atom scavenged three Na atoms (Farges and Brown 1997), at 6.25 mol% TiO2, Si4+ in NS8 glass would be completely in Q4. In our system, there is no indication that the 29Si Q4 peak grows at the expense of the Q3 peak, requiring that Na–Ti bonding is minimal to absent along the NS4 and NS8 TiO2 glasses. It is noted that the previous research that indicated the presence of Na-titanite complexes (Farges et al. 1996a) was performed on more Na-rich (e.g., NS2) glasses. These glasses are in Na-silicate and Na-Ti-silicate liquidus fields (Fig. 1), as opposed to the SiO2 and TiO2 liquidus fields of NS4 and NS8 glasses. In these systems where Na activity is higher, it would be expected that Na-titanite complexes could form in the melt as suggested in Fig. 1.
In the case of NS4 and NS8 titanium bearing glasses, the presence of five coordinated Ti could be incorporated via a charge-balancing mechanism that does not rely on Na-titanate clusters. For example, titanium complexes with both 3 and 2 coordinated oxygens, [5]Ti4+ + 3[3]O−3/2 + 2[2]O−1. In this case, Ti would form [5]Ti clusters at low concentrations through sharing of [3]O between 3 [5]Ti atoms, and [2]O could interact with [5]Ti, [4]Si, or [4]Ti. This mechanism helps to explain both the apparent shift in Q4 (as opposed to diminution of Q3) peaks for the 29Si observed in the NMR spectra as well as the initial pure [5]Ti observed in the Ti XAFS pre-edge region. This type of [5]Ti bonding accounts for both the XAFS and NMR observations, and suggests that even at low bulk Ti content, [5]Ti is bonded primarily through clustering mechanisms with negligible Na–Ti interactions.
Because TiO2 is being added in excess to this system (as opposed to being added as an equimolar replacement of SiO2), we envision Ti exerting an effect similar to oxygen triclusters that have systematically been observed in peraluminous melts, where the amount of Al in the system is in excess of charge-balancing alkali cations (Stebbins et al. 2001; Toplis et al. 1997). Alternatively, if the influence of Ti on Si NMR is similar to that of Al in these glasses, it is possible that there is a diminution of the Q3 peak as a result of a concomitant growth of a Q4 (Ti = 2) peak. Although we do not this this is the most likely scenario given the data collected, it is difficult to specifically discount this mechanism given the broadness of the peaks at increasing bulk TiO2 content.
The 29Si NMR data indicate that as Ti concentration increases, incorporation of [4]Ti occurs predominantly through the interaction of Ti with Q4 Si, as a network-forming cation in Q4 speciation or as will be shown later as isolated Ti clusters. In Fig. 7, three [4]Ti4+ substitution schemes are considered. Naturally, this figure is not meant to represent all Ti in the system (most Ti is in five-fold coordination), but rather to address potential substitution mechanisms for [4]Ti4+. In Fig. 7a, Ti4+ substitution for Si4+ in a Q3 species with the neighboring sodium cation providing charge balance is envisioned. From the perspective of the 29Si NMR spectra, where this substitution to be significant, one would observe a reduction in Q3 intensity and an increase in Q4 intensity, where such spectral changes are not observed in either Fig. 3 or 4.

Three different mechanisms by which [4]Ti can be incorporated into the glass structure include: a in replacement of Q3 Si, which would induce a loss of Q3 Si and growth of Q4 Si in the NMR spectra, which is not observed, b for Q4 Si in the network with no adjacent Q3 Si, which would incite the growth of an additional Q4 peak at a slightly higher energy than the primary Q4 peak, and c for Q4 Si adjacent to Q3 Si, which would incite a peak shift toward higher frequency of the Q3 Si peak. The latter is not observed in the NS8 glasses, but a slight shift in peak position is detectable in the NS4 glasses (Fig. 3)
In the scenario where Ti4+ substitutes for Si4+ in Q4 species (Fig. 7b), from a perspective of 29Si NMR, one would expect to see a shift in Q4 to a higher frequency if the effect of titanium coordination is similar to the case of Al3+ substitution into Si4+ Q4 species (Fig. 2). This shift is the result of the growth of a new Q4 peak at the expense of the original Q4 peak, we expect this new Q4 (Ti = 1) peak to occur at a slightly higher frequency of the Q4 (Ti = 0) peak similar to the effect of Al substitution (Fig. 2). In both NS4 and NS8 (with TiO2), the 29Si NMR frequency shift of the “Q4” peak supports the scenario that [4]Ti4+ substitutes for Si4+ in Q4 species. Note that in Fig. 7c, a Ti4+ substitution into a Q4 species adjacent to a Q3 species would also be expected to impart a small positive frequency shift in 29Si Q3; however, no such shift (as is observed with Q4) is evident for the NS8 + TiO2 glasses, but in the case of NS4 + TiO2, there is possibly evidence of the growth of very weak signal at frequencies at and above − 80 ppm, potentially suggesting the growth of a broad and very weak peak buried below the prominent Q3 peak as will be considered below.
For the purpose of completeness, we note that it is conceivable that a mechanism might exist wherein titanium solution into silicate rich (Q4) domains draws silica away from Q3 domains, which would necessitate the formation of Q2 species. In the present case, there is no 29Si NMR evidence for any development of Q2 (Fig. 2) and, therefore, there is no evidence to support this potential scenario for these melt (glass) compositions. It is noted that in a Raman study along the NS2-NT2 join (Mysen and Neuville 1995) that the silicate NBO increased as NT2 increased implying behavior that is consistent what is observed for the NS4-TiO2 and NS8-TiO2 glasses studied here.
Based on the 29Si NMR data presented in Figs. 3 and 4, it therefore appears that the most conservative interpretation is that for high silica content sodium silicate melts (e.g., NS4 and NS8 analyzed as glasses), addition of [4]TiO2 predominantly perturbs the silicate rich (Q4) domains and minimally (if at all) perturbs the sodic rich domains (Q3). Given that NS8 has 3 times more Q4 relative to Q3 than NS4, it is perhaps not surprising that the magnitude of the perturbation to 29Si NMR is less in NS8 glasses with equivalent increases in TiO2 (see Figs. 3 and 4).
The 29Si NMR spectra in Figs. 3 and 4 are quite broad at higher Ti compositions and there are little to no constraints to perform robust spectral fitting—this is particularly the case for NS4-Ti (Fig. 3). Therefore, instead of fitting the spectral data, we use the known effect of Al3+ substitution on Si Q4 frequency as a guide (Fig. 2) (Lippmaa et al. 1981) for what Ti4+ would be expected to do, i.e., cause the formation of a new peak at ~ 100 ppm due to Q4 (Ti = 1) species. We then explore how the 29Si spectrum could be progressively perturbed with increased titanium addition by forward modeling the growth of this new Q4 (Ti = 1) peak with proportional loss of Q4 (Ti = 0) peak intensity. We test the validity of the forward models by whether these model spectra replicate what is observed in the actual data. As noted previously and shown in Fig. 2, the 29Si peak for Si Q4 with one aluminum nearest neighbor (Q4 Al = 1) lies between NS4 and NS8 Q3 and Q4 (at 0% TiO2) for both NS4 and NS8. For modeling purposes, we assume that the peak for Q4 (Ti = 1) also lies at between Q3 (Ti = 0) and Q4 (Ti = 0) based on the idea that the similarity of electronegativity with Ti4+ and Al3+ will result in a similar chemical shift.
In Fig. 8a and b, model spectra of NS4 and NS8 glasses are presented using identical Gaussian peak shapes and fixed frequencies for the Q3 (Ti = 0), Q4 (Ti = 0), and Q4 (Ti = 1) peaks. Figure 8c and d shows a spectral deconvolution of these peaks at 40% (by area) Q4 (Ti = 1) and 60% Q4 (Ti = 0). With titanium addition, we assume that silicon Q4 species are systematically transformed from having 0 neighboring “Ti4+” (Q4 Ti = 0) up to 100% Q4 with one neighboring “Ti4+” (Q4 Ti = 1). Note that for NS4 with 100% Q4 (Ti = 1), the corresponding total titanium concentration would be 6.25 mol% TiO2 (e.g., 1 Ti for every 6 Q4 Si), for the more silica rich (and Q4 rich) NS8, the corresponding total concentration of titanium at 100% Q4 (Ti = 1) would be 9.5 mol% TiO2. For titanium concentrations above these values, either Q4 (Ti = 2) species would have to form which by comparison with Q4 (Al = 2) (Fig. 2) would lead to an apparent increase in intensity around Q3 (Ti = 0), or Q3 (Ti = 1) would have to form which by comparison with the effects of aluminum would lead to the prediction of increased intensity at ~− 80 ppm. If TiO2 begins to cluster into titanium oxide rich domains, the presence of such would not be predicted to influence or perturb the 29Si NMR spectra.

Simulated 29Si NMR spectra for Ti-bearing Na-silicate glasses, generated using Gaussian peaks at the positions and shapes of the Ti-free a NS4 and b NS8 glasses, by varying the proportions of Q4 (Ti = 1) and Q4 (Ti = 0) peaks. The peaks that compose these model spectra at 40% Q4 (Ti = 1) and 60% Q4 (Ti = 0) are shown in 8 c and 8 d. Increasing Ti content in the simulations is represented by the growth of a third Q4 peak at a higher frequency as a result of nearest-neighbor interactions with [4]Ti nearest neighbors. The simulations are similar to the actual spectra, with the notable difference that the simulations over-estimate the amount of Q4 Si with [4]Ti nearest neighbors Q4 (Ti = 1). e, f If Ti was incorporated entirely by the Q4 (Ti = 1) mechanism, then observed spectra would be reflective of 100% Q4 (Ti = 1) at high Ti content in NS4 and NS8 glasses. However, NS8 glass with 15 mol% TiO2 and NS4 glass with 10 and 15% TiO2 which would have all Si in Q4 (Ti = 1) at these concentrations are still significantly far from Q4 (Ti = 1). This over-estimation suggests that Ti is clustering at relatively low bulk TiO2, an observation that is supported by the Ti XAFS observations of significant [5]Ti in the NS8 glasses
The forward model of the NS4-TiO2 system (Fig. 8a) is similar to the measured 29Si NMR data (Fig. 3) at low Q4 (Ti = 1) concentrations where a progressive shift of Q4 to slightly higher frequency occurs with titanium addition. Interestingly, the actual 29Si spectra for NS4 with 15% Ti (Fig. 3) looks most similar to model at 60–70% Q4 (Ti = 1) (Fig. 8a). This suggests Si–Ti interactions saturate with Q4 (Ti = 1) at ~ 4–5 mol% Ti, suggesting that the remaining (~ 10 mol% at 15 mol% TiO2 oxide) Ti4+ forms oxide clusters whose size and number increase up to the maximum titanium concentration without further perturbation to the 29Si spin system.
The forward model of the NS8-TiO2 system (Fig. 8b) is also similar to the measured 29Si NMR data (Fig. 4) at low Q4 (Ti = 1) concentrations. At 100% Q4 (Ti = 1), the apparent shift in “Q4” exceeds what is observed in the actual NS8 29Si NMR spectra (Fig. 4). At 50% Q4 (Ti = 1), one observes a model spectrum that is very similar to that of NS8 with 15 mol% TiO2 (Fig. 8b). As is the case with the NS4-TiO2 system, this suggests that Si–Ti interactions saturate with Q4 (Ti = 1) again at 4–5 mol% TiO2. Again, at 15 mol% TiO2, the remaining ~ 10 mol% TiO2 must reside in Ti oxide clusters whose presence would not be evident in the 29Si NMR spectra (Figs. 3 and 4).
The analyses of the forward models in comparison with the actual 29Si NMR spectra (Fig. 8e, f) indicate that the perturbation to silicate is much less than what is possible given the amount of TiO2 available. In both the case of NS4 and NS8 TiO2 series, the principle perturbation due to TiO2 addition is the formation of a modest amount of Q4 (Ti = 1) groups. It is noted that the Pauling radii of Si4+ is 41 pm, Al3+ is 50 pm, and Ti4+ is 68 pm (Pauling 2014). The much larger radius of Ti4+ relative to Si4+ may limit titanium substitution to Si4+ tetrahedral to no more than one Ti4+ [e.g., Q4 (Ti = 1)]. The 29Si NMR spectra indicate that titanium oxide also likely begins to cluster with itself even at relatively low bulk titanium concentrations (Farges 1999; Kim et al. 2000). Obviously, the 29Si NMR spectra only report on what directly perturbs silicon, so as titanium oxide begins to cluster with titanium oxide; such clustering will “dilute” the perturbation of the silicate component of the melt/glass and will minimize what is observed with 29Si solid state NMR. Large Ti clusters would likely result in the formation of higher order Ti coordination (octahedral Ti) that is not observed, suggesting the clustering occurs in small localized regions dispersed throughout the glass matrix. However, these regions also must be large enough that no significant Q4 (Ti = 2) or Q4 (Ti = 3) peaks are generated at high bulk TiO2 content as a result of Ti dispersal between SiO2 tetrahedra. The fact that there is no evidence for Q3 (Ti = 1) in the 29Si NMR spectra suggests that the Q4 (Ti = 1) species may signify the interface between alkali silicate oxide phases and titanium oxide clusters.
Conclusions, implications, and importance for natural systems
This work highlights the complexity of Ti incorporation in silicate glasses, and the variable mechanisms by which Ti can be incorporated into melts. In natural magmatic liquids, changing solubility mechanisms could significantly influence the stability of Ti-rich phases and the calculation of Ti4+ activity in rutile-undersaturated melts (Ghiorso and Gualda 2013). A better understanding of how Ti4+ is incorporated into glasses can also help explain the physical phenomena (e.g., compressibility, tensile strength) that occur as Ti4+ is incorporated into glass compositions (Scannell et al. 2016).
Although the observations presented here are not directly applicable to natural silicate glasses (e.g., rhyolites, basalts), they provide insight into important processes operating in natural systems. In particular, the observations shown here clearly demonstrate a change in the solubility mechanism for Ti4+ in glasses with changes in the bulk TiO2 content of the glass. Thermodynamically, changes in the solubility mechanism of Ti4+ in glasses will change the activity coefficient for the TiO2 fusion reaction as a function of TiO2 content. In turn, this will play a significant role in the calculation of Ti4+ activity in silicate melts. Because accurate estimation of Ti4+ activity is required for numerous applications (phase stability, trace-element thermobarometry), it is important for similar observations to be made on natural systems to gain a more complete picture of the solubility mechanisms for Ti4+ in melts and glasses. Specifically, changes in solubility mechanisms could result in the overestimation of TiO2 activity in rutile-undersaturated melts using rutile-saturation modeling (Ackerson and Mysen in press). Employing NMR in combination with Ti K-edge XAFS for other model systems could provide specific evidence to help explain the anomalous behavior of Ti4+ in glasses.
The applicability of the combined use of these two techniques will be diminished in systems (e.g., rhyolites), where the Ti saturation concentration at magmatic temperatures is too low to exert meaningful or detectible influence on 29Si NMR spectra. However, these combined techniques could be useful in understanding how Ti4+ is incorporated into more mafic melts, where Ti4+ saturation concentrations are significantly higher (Gaetani et al. 2008). Utilizing these techniques could shed light on rutile solubility in mafic systems, inform our understanding of the processes leading to Nb-Ta anomalies, and bolster our understanding of the origins of high-Ti lunar glasses (Van Orman and Grove 2000).
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article (and its additional files).
References
Ackerson M, Mysen BO. Experimental observations of TiO2 activity in rutile-undersaturated melts. Am Mineral in press
Ackerson MR, Mysen BO, Tailby ND, Watson EB (2018) Low-temperature crystallization of granites and the implications for crustal magmatism. Nature 559(7712):94–97
Ackerson MR, Tailby ND, Watson EB (2017) XAFS spectroscopic study of Ti coordination in garnet. Am Mineral 102(1):173–183
Cody GD, Mysen BO, Lee SK (2005) Structure vs. composition: a solid state 1H and 29Si NMR study of quenched glasses along the Na2O-SiO2-H2O join. Geochim Cosmochim Acta 69:2373–2384
Farges F (1997) Coordination of Ti4+ in silicate glasses: a high-resolution XANES spectroscopy study at the Ti K edge. Am Mineral 82:36–43
Farges F (1999) A Ti K-edge EXAFS study of the medium range environment around Ti in oxide glasses. J Non-Cryst Solids 244:25–33
Farges F, Brown GE (1997) Coordination chemistry of titanium (IV) in silicate glasses and melts: IV. XANES studies of synthetic and natural volcanic glasses and tektites at ambient temperature and pressure. Geochim Cosmochim Acta 61(9):1863–1870
Farges F, Brown GE Jr, Navrotsky A, Gan H, Rehr JJ (1996a) Coordination chemistry of Ti(IV) in silicate glasses and melts: II. Glasses at ambient temperature and pressure. Geochim Cosmochim Acta 60(16):3039–3053
Farges F, Brown GE Jr, Rehr JJ (1996b) Coordination chemistry of Ti(IV) in silicate glasses and melts: I. XAFS study of titanium coordination in oxide model compounds. Geochim Cosmochim Acta 60(16):3023–3038
Farges F, Lefrère Y, Rossano S, Berthereau A, Calas G, Brown GE Jr (2004) The effect of redox state on the local structural environment of iron in silicate glasses: a combined XAFS spectroscopy, molecular dynamics, and bond valence study. J Non-Cryst Solids 344(3):176–188
Gaetani GA, Asimow PD, Stolper EM (2008) A model for rutile saturation in silicate melts with applications to eclogite partial melting in subduction zones and mantle plumes. Earth Planet Sci Lett 272(3–4):720–729
Ghiorso MS, Gualda GAR (2013) A method for estimating the activity of titania in magmatic liquids from the compositions of coexisting rhombohedral and cubic iron–titanium oxides. Contrib Mineral Petrol 165(1):73–81
Glasser FP, Maar J (1979) Phase relations in the system Na2O-TiO2-SiO2. J Am Ceram Soc 62:42–47
Hayden LA, Manning CE (2011 May) Rutile solubility in supercritical NaAlSi3O8–H2O fluids. Chem Geol 284(1–2):74–81
Hayden LA, Watson EB (2007) Rutile saturation in hydrous siliceous melts and its bearing on Ti-thermometry of quartz and zircon. Earth Planet Sci Lett 258(3–4):561–568
Henderson GS, Fleet ME (1995) The structure of Ti-silicate glasses by microRaman spectrocopy. Can Mineral 33:399–408
Janes N, Oldfield E (1985) Prediction of silicon-29 nuclear magnetic resonance chemical shifts using a group electronegativity approach: applications to silicate and aluminosilicate structures. Adv Ceram Mater 107(24):6769–6775
Kim WB, Choi SH, Lee JS (2000) Quantitative analysis of Ti-O-Si and Ti-O-Ti bonds in Ti-Si binary oxides by the linear combination of XANES. J Phys Chem B 104(36):8670–8678
Kuemmerlen J, Merwin LH, Sebald A, Keppler H (1992) Structural role of water in sodium silicate glasses: results from silicon-29 and proton NMR spectroscopy. J Phys Chem 96(15):6405–6410
Lange RA, Navrotsky A (1993) Heat capacities of TiO2-bearing silicate liquids: Evidence for anomalous changes in configurational entropy with temperature. Geochim Cosmochim Acta 57(13):3001–3011
Le Losq C, Mysen BO, Cody GD (2015) Water and magmas: insights about the water solution mechanisms in alkali silicate melts from infrared, Raman, and 29Si solid-state NMR spectroscopies. Prog Earth Planet Sci 2(1) Available from: http://www.progearthplanetsci.com/content/2/1/22. Cited 2015 Aug 25
Lippmaa E, Maegi M, Samoson A, Engelhardt G, Grimmer AR. Structural studies of silicates by solid-state high-resolution silicon-29 NMR. 2002. Available from: https://pubs.acs.org/doi/abs/10.1021/ja00535a008. Cited 2019 Sep 26
Lippmaa E, Maegi M, Samoson A, Tarmak M, Engelhardt G (1981) Investigation of the structure of zeolites by solid-state high-resolution silicon-29 NMR spectroscopy. J Am Chem Soc 103(17):4992–4996
Liska M, Simurka P, Antalik J, Perichta P (1996) Viscosaity of titania-bearing sodium silicate melts. Chem Geol 128:199–206
Liu Q, Lange RA (2001) The partial molar volume and thermal expansivity of TiO2 in alkali silicate melts: systematic variation with Ti coordination. Geochim Cosmochim Acta 65:2379–2394
Maekawa H, Maekawa T, Kawamura K, Yokokawa T (1991) 29Si MAS NMR investigation of the Na2O-Al2O3-SiO2 glasses. J Phys Chem 95(18):6822–6827
Magi M, Lippmaa E, Samoson A, Engelhardt G, Grimmer AR (1984) Solid-state high-resolution silicon-29 chemical shifts in silicates. J Phys Chem 88(8):1518–1522
Mauri F, Pasquarello A, Pfrommer BG, Yoon Y-G, Louie SG (2000) Si-O-Si bond-angle distribution in vitreous silica from first-principles ${}^{29}\mathrm{Si}$ NMR analysis. Phys Rev B 62(8):R4786–R4789
Morsi MM, El-Shennawi AWA (1984) Some physical properties of silicate glasses containing TiO2 in relation to their structure. Phys Chem Glasses 25:64–68
Mysen B, Neuville D (1995) Effect of temperature and TiO2 content on the structure of Na2Si2O5Na2Ti2O5 melts and glasses. Geochim Cosmochim Acta 59(2):325–342
Mysen BO, Kumamoto K, Cody GD, Fogel ML (2011 Oct) Solubility and solution mechanisms of C–O–H volatiles in silicate melt with variable redox conditions and melt composition at upper mantle temperatures and pressures. Geochim Cosmochim Acta 75(20):6183–6199
Mysen BO, Richet P (2005) Silicate glasses and melts: properties and structure. Elsevier, Amsterdam; Boston Available from: http://public.eblib.com/choice/publicfullrecord.aspx?p=270091. Cited 2016 Nov 16
Newville M (2013) Larch: an analysis package for XAFS and related spectroscopies. J Phys Conf Ser 430:012007
Pauling L (2014) General chemistry. Courier Corporation
Romano C, Paris E, Poe BT, Giuli G, Dingwell DB, Mottana A (2000) Effect of aluminum on Ti-coordination in silicate glasses: a XANES study. Am Mineral 85:108–117
Roskosz M, Mysen BO, Cody GD (2006) Dual speciation of nitrogen in silicate melts at high pressure and temperature: an experimental study. Geochim Cosmochim Acta 70(11):2902–2918
Roskosz M, Toplis MJ, Richet P (2004) The structural role of Ti in aluminosilicate liquids in the glass transition range: insights from heat capacity and shear viscosity measurements. Geochim Cosmochim Acta 68(3):591–606
Ryerson FJ, Watson EB (1987) Rutile saturation in magmas: implications for TiNbTa depletion in island-arc basalts. Earth Planet Sci Lett 86(2–4):225–239
Scannell G, Barra S, Huang L (2016) Structure and properties of Na2O-TiO2-SiO2 glasses: role of Na and Ti on modifying the silica network. J Non-Cryst Solids 448:52–61
Scannell G, Huang LP, Rouxel T (2015) Elastic properties and indentation cracking behavior of Na2O-TiO2-SiO2 glasses. J Non-Cryst Solids 429:129–142
Smith JV, Blackwell CS (1983 May) Nuclear magnetic resonance of silica polymorphs. Nature 303(5914):223–225
Stebbins JF, Oglesby JV, Kroeker S (2001) Oxygen triclusters in crystalline CaAl4O7 (grossite) and in calcium aluminosilicate glasses: 17O NMR. Am Mineral 86(10):1307–1311
Tang M, Lee C-TA, Chen K, Erdman M, Costin G, Jiang H (2019) Nb/Ta systematics in arc magma differentiation and the role of arclogites in continent formation. Nat Commun 10(1):1–8
Toplis MJ, Dingwell DB, Lenci T (1997 Jul 1) Peraluminous viscosity maxima in Na2OAl2O3SiO2 liquids: the role of triclusters in tectosilicate melts. Geochim Cosmochim Acta 61(13):2605–2612
Van Orman JA, Grove TL (2000) Origin of lunar high-titanium ultramafic glasses: constraints from phase relations and dissolution kinetics of clinopyroxene-ilmenite cumulates. Meteorit Planet Sci 35(4):783–794
Watson EB, Harrison TM (2005) Zircon thermometer reveals minimum melting conditions on earliest earth. Science 308(5723):841–844
Waychunas GA (1987) Synchrotron radiation XANES spectroscopy of Ti in minerals: effects of Ti bonding distances, Ti valence, and site geometry on absorption edge structure. Am Mineral 72:89–101
Xiong X-L (2006) Trace element evidence for growth of early continental crust by melting of rutile-bearing hydrous eclogite. Geology 34(11):945–948
Yarker CA, Johnson PAV, Wright AC, Wong J, Greegor RB, Lytle FW et al (1986) Neutron diffraction and EXAFS evidence for TiO5 units in vitreous K2O.TiO2.2SiO. J Non-Cryst Solids 79:7–136
Zotov N, Keppler H (1998) The influence of water on the structure of hydrous sodium tetrasilicate glasses. Am Mineral 83(7–8):823–834
Acknowledgements
We thank the editorial staff for handling of this manuscript, and Megan Holycross, Tony Lanzirotti, Matt Neuville, and Steve Sutton for the help in collecting Ti EXAFS data. Thanks also to two anonymous reviewers whose insights helped improve the clarity of the manuscript. We gratefully acknowledge the W. M. Keck Foundation and the NSF MRI program for the support of the W. M. Keck Solid State NMR facility at the Geophysical Laboratory. This research was also supported by the Carnegie Institution for Science’s Postdoctoral Fellowship Program.
Funding
This research was funded through the Carnegie Institution’s Postdoctoral Fellowship Program, which contributed research and stipend support for the lead author during the completion of this project. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOW Office of Science by Argonne National Laboratory under Contract no. DE-AC02-06CH11357, which enabled collection of the XANES data reported herein.
Author information
Authors and Affiliations
Contributions
MA carried out the experimental synthesis and EXAFS analyses, GC performed the NMR experiments, and BM contributed to the structural interpretations. All authors contributed to the intellectual content of the manuscript, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Figure S1.
Ti XANES spectrum of an NS8 glass with 15 mol. % TiO2, compared with a spectrum from [6]Ti-bearing crystalline titanite (CaTiSiO5). The sharp white line peak in the titanite spectrum above the absorption edge is an indicator of the crystalline nature of the titanite. The broad edge feature in the Ti15NS8 spectrum suggests the material is amorphous. Supplementary Figure S2. Ti XANES spectrum of NS8 glasses with 10 mol % TiO2 synthesized from 1186, 1250, and 1580 °C. All glasses produced near-identical spectra, suggesting that there is no effect of dwell temperature on the solubility mechanism of Ti in NS8 glasses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ackerson, M.R., Cody, G.D. & Mysen, B.O. 29Si solid state NMR and Ti K-edge XAFS pre-edge spectroscopy reveal complex behavior of Ti in silicate melts. Prog Earth Planet Sci 7, 14 (2020). https://doi.org/10.1186/s40645-020-00326-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40645-020-00326-2