
Abstract
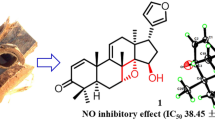
A new havanensin-type limonoid, 16β-hydroxydysobinin (1), along with four known limonoids (2–5), have been isolated from the seeds of Chisocheton macrophyllus. The chemical structure of the new compound was determined by referencing spectroscopic data, and by comparison to those related spectra previously reported. Each compound was evaluated for their cytotoxic effects against Michigan Cancer Foundation-7 (MCF-7) breast cancer cells and display no significant activity.
Similar content being viewed by others

Introduction
Limonoids, known as degraded triterpenes, are derived from a precursor with a 4,4,8-trimethyl-17-furanylsteroid four-ring skeleton labelled as A, B, C and D rings [1]. Limonoids are a class of secondary metabolites found in the order Rutales and the Meliaceae and Rutaceae family [2]. Meliaceae is a family of timber trees that are a rich source for limonoids and are widely distributed in tropical and subtropical regions with 50 genera and more than 1400 species [3, 4]. Limonoids isolated from species of the family Meliaceae have been reported to have biologically activity as antifeedant, antimicrobial, antimalarial, and cytotoxins [5,6,7,8]. Genus Chisocheton is the second largest in the family Meliaceae, consisting of more than 50 species distributed across Nepal, India, Bhutan, Myanmar, South China, Thailand, Indonesia, Malaysia, and Papua New Guinea [7, 9].
Chisocheton macrophyllus is a species distributed in the Nicobar Islands, peninsular Thailand, peninsular Malaysia, Singapore, Sumatera, Anambas Islands, Java and Borneo Islands [10]. Its seeds have been reported to yield bioactive limonoids such as dysobinol, 7α-hydroxyneotricilenone, dysobinin and nimonol with cytotoxic activity against P-388 murine leukemia cells [11], whereas the leaves to yield Epstein-Barr virus activation of Triterpenoids [12]. After further investigations for cytototoxic limonoids from the seeds of C. macrophyllus, we found and structural elucidation of a new havenensin-type limonoids (1) and four known limonoids (2–5), along with their cytotoxic activity against MCF-7 breast cancer cells. Herein, the isolation, structural elucidation and cytotoxic activity against MCF-7 breast cancer cells are discussed.
Materials and methods
Plant materials
Seeds of C. macrophyllus were collected from Bogor Botanical Garden, Bogor, West Java Province, Indonesia. The plant was identified by Mr. Harto, the staff of Bogoriense Herbarium, Research Center for Biology, Indonesia Science Institute, Bogor, Indonesia and a voucher specimen (No. Bo-1295453) was deposited at the Herbarium.
Instruments and reagents
Optical rotations were measured on a Perkin Elmer 341 Polarimeter (Waltham, MA, USA). UV spectra was measured using a TECAN Infinite M200 pro with MeOH. Furthermore, the IR spectra and mass spectra were recorded on a One PerkinElmer spectrum-100 FT-IR in KBr and Waters Xevo QTOF MS, respectively. NMR spectra were obtained with Bruker Topspin at 500 MHz for 1H and 125 MHz for 13C (compound 1) and for compounds 2–5 using JEOL JNM-ECZ500R/S1 at 500 MHz for 1H and 125 MHz for 13C, using tetra methylsilane (TMS) as the internal standard. Chromatographic separations were carried out on the silica gel 60 (70–230 and 230–400 mesh, Merck). Thin layer chromatography (TLC) analysis was carried out on 60 GF254 (Merck, 0.25 mm) using various solvent systems, and measured by irradiation under ultraviolet–visible light Vilber Lourmat (λ 254 nm dan 365 nm) followed by heating of silica gel plates, sprayed with 10% H2SO4 in ethanol and Ehrlich’s reagent (p-Dimethylaminobenzaldehyde in ethanol).
Extraction and isolation of C. macrophyllus
The dried and powdered seeds of C. macrophyllus (2.5 kg) were extracted with methanol at room temperature for 3 days (3 × 5 L). After removal of the solvent under a vacuum, a total of 360 g of methanol extract was obtained and partitioned with n-hexane (3 × 3 L), ethyl acetate (3 × 2 L) and n-butanol (3 × 2 L). Evaporation resulted in crude extracts of n-hexane (146.6 g), ethyl acetate (60.8 g) and n-butanol (14.6 g) respectively. The n-hexane soluble fraction (140 g) was subjected to vacuum-liquid chromatography (VLC) column packed with silica gel 60 using a gradient of n-hexane, ethyl acetate and methanol (10% stepwise) to afford thirteen fractions (A-M). Fraction D (5.4 g) was subjected to silica gel column chromatography using a gradient of n-hexane and ethyl acetate (5% stepwise) as eluting solvent to afford five subfractions (D1-D5). Subfraction D2 (165.7 mg) was chromatographed on a column of silica gel eluted with n-hexane: dichloromethane: ethyl acetate (2:7.5:0.5) to give 1 (15.3 mg). Fraction F (4.4 g) was subjected to a silica gel column chromatography using a gradient of n-hexane and ethyl acetate (5% stepwise) as eluting solvent to afford twelve subfractions (F1-F12). Subfraction F5 (1.2 g) was chromatographed on a column of silica gel eluted with n-hexane: dichloromethane: ethyl acetate (2:7.5:0.5) to give 3 (19.7 mg) and four subfractions (F5A-F5D). Furthermore, subfraction F5D (308.3 mg) was chromatographed on a column of silica gel eluted with n-hexane: dichloromethane: ethyl acetate (1:8.5:0.5) to give 2 (12.8 mg). Fraction H (1.8 g) was subjected to a silica gel column chromatography using a gradient of n-hexane and ethyl acetate (5% stepwise) as eluting solvent to give 5 (12.0 mg). Fraction J (1.5 g) was subjected to a silica gel column chromatography using a gradient of n-hexane and ethyl acetate (5% stepwise) to afford nineteen subfractions (J1-J19). Subfraction J9 (50.3 mg) was chromatographed on a column of silica gel eluted with n-hexane: dichloromethane: ethyl acetate (4:5.5:0.5) to give 4 (3.0 mg).
16β-hydroxydysobinin (1): Colorless needle crystals; mp: 205–207 °C; [\(\alpha\)]27D \(+1\)22.5° (c 0.2, MeOH); UV (MeOH) λmax 284 nm; IR (KBr) vmax 3509, 2929, 1744, 1670, 1502, 1366, 1386, 1248 cm−1; HR-TOFMS m/z 511.2634 [M + H]+, (calcd. for C30H39O7 m/z 511.2696); 1H-NMR (CDCl3, 500 MHz) and 13C-NMR (CDCl3, 125 MHz) see Table 1.
Cytotoxic activity test
The cytotoxicity of compounds 1–5 was determined with a cell viability test using PrestoBlue® assay. The cells were maintained in a Roswell Park Memorial Institute (RPMI) medium with 10% (v/v) Fetal Bovine Serum (FBS) and 1 \(\upmu\)L/1 mL antibiotics (1% Penicillin–Streptomycin). Cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2. MCF-7 cells plated in 96 multiwell culture plates at a density of 1.7 \(\times\) 104 cells/well. After twenty-four hours, the medium was discarded and fresh medium containing sample with different concentrations 7.81, 15.63, 31.25, 62.50, 125.00, 250.00, 500.00, 1000.00 μg/mL and control was added. After incubation with the sample for 24 h, PrestoBlue® reagent (resazurin dye) was added. The PrestoBlue® assay results were read using a multimode reader at 570 nm. The IC50 values were determined by linier regression method using Microsoft Excel software. The IC50 value corresponds to the concentration of compounds that decreases by 50% the number of viable cells and the absorbance in control corresponds to 100% viability.
Results and discussion
The n-hexane fraction from the seeds of C. macrophyllus was subjected to vacuum-liquid chromatography (VLC) column packed with silica gel 60 by gradient elution. The VLC fractions were repeatedly subjected to normal phase column chromatography on silica gel to yield compounds 1–5 (Fig. 1).

Structures of compounds 1–5
Compound 1 was isolated as colorless needle crystals. The molecular formula was determined to be C30H39O7 based on the high resolution time-of-flight mass spectrometry (HR-TOFMS) spectra (Additional file 1; Fig. S8) at m/z 511.2634 [M + H]+ (calcd. for C30H39O7 m/z 511.2696) and nuclear magnetic resonance (NMR) data (Table 1), indicating the presence of twelve degrees of unsaturation. The ultraviolet (UV) spectrum showed maximum absorption at 284 nm, indicating the presence of an \(\alpha ,\beta\)-unsaturated ketone [13, 14]. Infrared (IR) absorptions spectra suggested the presence of hydroxyl (3509 cm−1), aliphatic (2929 cm−1), carbonyl ester (1744 cm−1), \(\alpha ,\beta\)-unsaturated carbonyl (1670 cm−1), olefinic (1502 cm−1), gem-dimethyl (1366 and 1386 cm−1) and ether groups (1248 cm−1).
The 1H-NMR spectrum (Additional file 1; Fig. S1) showed five tertiary methyls at δH 0.95 (3H, s, Me-28), 1.20 (3H, s, Me-29), 1.28 (3H, s, Me-19), and 1.33 (6H, s, Me-18 and Me-30) as well as two acetoxyl groups at δH 2.04 (3H, s, Me-1′) and 2.07 (3H, s, Me-1″). In addition, three oxygenated protons δH 5.46 (1H, m, H-6), 5.41 (1H, d, J = 2.6 Hz, H-7) and 4.49 (1H, t, J = 7.7 Hz, H-16), a β-furan moiety δH 6.30 (1H, d, J = 1.45 Hz, H-22), 7.28 (1H, s, H-21), and 7.40 (1H, d, J = 1.45 Hz, H-23) and three olefinic protons at δH 5.48 (1H, d, J = 7.7 Hz, H-15), 5.88 (1H, d, J = 10.5 Hz, H-2) and 8.32 (1H, d, J = 10.5 Hz, H-1) were also observed in the 1H-NMR spectrum. The 13C NMR (Additional file 1; Fig. S2) along with distortions enhancement by polarization transfer (DEPT) (Additional file 1; Fig. S3) and heteronuclear single quantum coherence (HSQC) spectra (Additional file 1; Fig. S4) showed thirty carbons consisting of an \(\alpha\),\(\beta\)-unsaturated carbonyl at δC 204.4 (C-3), two acetoxyl groups at δC 21.3 (C-1′), 170.1 (C-2′), 22.0 (C-1″) and 170.3 (C-2″) and five methyls at δC 20.5 (Me-28), 20.6 (Me-29), 20.9 (Me-30), 28.7 (Me-18) and 31.6 (Me-19). The spectra also showed two methylene carbons at δC 34.3 (C-11) and 46.4 (C-12), three sp3 methine carbons at δC 45.4 (C-9), 47.5 (C-5) and 51.2 (C-17), four sp2 methine carbons at δC 110.9 (C-22), 119.3 (C-15), 124.5 (C-2) and 158.1 (C-1), three oxygenated sp3 methine carbons at δC 67.2 (C-16), 69.7 (C-6) and 74.0 (C-7), two oxygenated sp2 methine carbons at δC 139.7 (C-21) and 142.7 (C-23), four sp3 quaternary carbons at δC 41.6 (C-8), 42.7 (C-10), 45.0 (C-4), and 46.8 (C-13) and two sp2 quaternary carbons at δC 124.0 (C-20) and 160.6 (C-14). These functionalities accounted for seven out of the twelve degrees of unsaturation, while the remaining five degrees of unsaturation corresponded to the pentacyclic limonoid structure [6, 11, 15, 16]. The NMR spectra data of 1 resembled those of previously reported dysobinin [16, 17], except for the appearance of oxygenated signals [δH 4.49 (1H, t, J = 7.7 Hz), δC 67.2], thus suggesting that 1 was a hydroxyl analog of dysobinin. Position of the hydroxyl group at C-16 was determined through the 1H-1H correlated spectroscopy (1H-1H COSY) and proton multiple bond connectivity (HMBC) experiments (Fig. 2, Additional file 1; Fig. S5 and Fig. S6). Correlations from methyl protons at δH 1.33 (CH3-18) to δC 51.2 (C-17), oxygenated sp3 methine at δH 4.49 (H-16) to δC 124.0 (C-20) and methyne proton δH 2.82 (H-17) to δC 139.7 (C-21) and δC 110.9 (C-22) were used to assign the hydroxyl group and a furan ring attached at C-16 and C-17, respectively.

Selected HMBC and 1H-1H COSY correlations for 1
Based on the 1H-1H COSY spectrum of 1, correlation in H1-H2, H5-H6-H7, H9-H11-H12, H16-H17 and H22-H23 supported the presence of a havanensin-type limonoid structure in 1 [15, 17, 18]. The HMBC spectrum showed 3 J correlations between sp2 methine proton signal δH 8.32 (H-1) to δC 47.5 (C-5) and carbonyl at δC 204.4 (C-3) and δH 5.88 (H-2) to δC 42.7 (C-10), indicating the presence of α, β-unsaturation ketone located at C-1, C-2 and C-3, respectively. Correlations from oxygenated sp3 methine protons at δH 5.46 (H-6) to δC 45.0 (C-4) and δC 170.1 (C-2′) and δH 5.41 (H-7) to δC 45.4 (C-9) as well as δH 2.04 (H-1′) to δC 170.1 (C-2′) and δH 2.07 (H-1″) to δC 170.3 (C-2″), indicate that an acetyl group was attached at C-6 and C-7, respectively.
The relative stereochemistry of hydroxyl group at C-16 of 1 was determined by a nuclear overhauser and exchange spectroscopy (NOESY) experiment (Fig. 3 and Additional file 1; Fig. S7). Comparison of oxygenated sp3 methine protons at δH 4.49 (H-16) and CH3-18 (δH 1.33) with \(\alpha\)-oriented, indicated that H-16 was \(\alpha\)-oriented and hydroxyl group at C-16 is \(\beta\)-oriented. Correlations between δH 5.41 (H-7) and CH3-30 (δH 1.33) with \(\beta\)-oriented, indicated that H-7 was \(\beta\)-oriented and acetyl group at C-7 is \(\alpha\)-oriented. Correlations between δH 5.46 (H-6) and CH3-19 (δH 1.28) with \(\beta\)-oriented, indicated that H-6 was \(\beta\)-oriented and acetyl group at C-6 is \(\alpha\)-oriented. Furthermore, the optical rotation of 1, [\(\alpha\)]27D \(+1\)22.5° (c 0.2, MeOH) is the same sign to those of previously reported dysobinin (6) ([α]20D + 150°) [17, 18]. Therefore, the structure of 1 was elucidated as the new havanensin-type of limonoid derivative and named 16β-hydroxydysobinin.

Selected NOESY correlations for 1
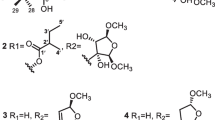
Four known compounds, 7-deacetylepoxyazadiradione (2), were previously synthesized as a derivative of epoxyazadiradione [19, 20], but isolated from a natural source for the first time. In addition, 6\(\mathrm{\alpha }\)-acetoxyepoxyazadiradione (3) and 6\(\mathrm{\alpha }\)-acetoxygedunin (4) [21] as well as dysobinin (5) [11, 17, 18] were identified by comparison of their spectroscopic data with previously reported values.
Cytotoxic activity
All isolated compounds were evaluated for the cytotoxic activity against MCF-7 breast cancer cell line and cisplatin is used as a positive control according to the method previously described [16, 22] and the results are shown in Table 2. Compound 1 showed the strongest activity against MCF-7 breast cancer cell with IC50 (inhibitory concentration, 50%) values of 45.91 \(\upmu\)M, suggesting that the presence of hydroxyl at C-16 can increase the cytotoxic activity. In addition, the presence of epoxy ring and ketone group, like in compound 2–4, showed weak activity, indicated the presence of epoxy ring and ketone group can decrease activity.
Change history
28 May 2023
Missing funding information has been added
References
Fang X, Di YT, Hao XJ (2011) The advances in the limonoid chemistry of the Meliaceae Family. Curr Org Chem 15:1363–1391
Li H, Peng Y, Zheng J (2016) (2016) Metabolic activation and toxicities of furanoterpenoids. AdvMolToxicol 10:55–97
Paritala V, Chiruvella KK, Thammineni C, Ghanta RG, Mohammed A (2015) Phytochemicals and antimicrobial potentials of mahogany family. Rev Bras Farmacogn 25:61–83
Tan QD, Luo XD (2011) MeliaceousLimonoids: chemistry and biological activities. Chem Rev 111:7437–7522
Yang MH, Wang JS, Luo JG, Wang XB, Kong LY (2009) Tetranortriterpenoids from Chisocheton paniculatus. J Nat Prod 72:2014–2018
Wong CP, Shimada M, Nagakura Y, Nugroho AE, Hirasawa Y, Taneda T, Awang K, Hadi AHA, Mohamad K, Shiro M, Morita H (2011) Ceramicines E-I, new limonoids from Chisocheton ceramicus. Chem Pharm Bull 59:407–411
Shilpi JA, Sahab S, Chong SL, Nahard L, Sarkerd SD, Awang K (2016) Advances in chemistry and bioactivity of the genus Chisocheton BLUME. Chem Biodiv 13:483–503
Chong SL, Hematpoor A, Hazni H, Azirun MS, Litaudon M, Supratman U, Murata M, Awang K (2019) Mosquito larvacidal limonoids from the fruits of Chisocheton erythrocarpus Hiern. Phytochem Lett 30:69–73
Katja DG, Farabi K, Nurlelasari HD, Mayanti T, Supratman U, Awang K, Hayashi H (2017) Cytototoxic constituents from the bark of Chisocheton cumingianus (Meliaceae). J Asian Nat Prod Res 6:1–5
Vossen VD and Umali BE (2002) Plant Resources of South East Asia, No. 14, Vegetable oil and fats, Prosea Foundation, Bogor, Indonesia.
Nurlelasari KDG, Harneti D, Wardayo MM, Supratman U, Awang K (2017) Limonoids from the seeds of Chisocheton macrophyllus. Chem Nat Compd 53:83–87
Inada A, Somekawa M, Murata H, Nakanishi T, Tokuda H, Nishino H, Iwashima A, Darnaedi D, Murata J (1993) Phytochemical studies on meliaceous plants. VIII structures and inhibitory effects on Epstein-Barr Virus activation of triterpenoids from leaves of Chisocheton macrophyllus KING. Chem Pharm Bull 41(3), 617–619.
Shiono Y, Miyazani N, Murayama T, Koseki T, Harizon KDG, Supratman U, Nakata J, Kakihara Y, Saeki M, Yoshida J, Uesugi S, Kimura K (2016) GSK-3β inhibitory activities of novel dichloresorcinol derivatives from Cosmospora vilior isolated from a mangrove plant. Phytochem Lett 18:122–127
Aisyah LS, Yun YF, Herlina T, Julaeha E, Zainuddin A, Nurfarida I, Hidayat AT, Supratman U, Shiono Y (2017) Flavonoid compounds from the leaves of Kalanchoe prolifera and their cytotoxic activity against P-388 murine leukimia cells. Nat Prod Sci 23(2):139–145
Najmuldeen IA, Hadi AHA, Mohamad K, Awang K, Ketuly KA, Mukhtar MR, Taha H, Nordin M, Litaudon M, Gueritte F, Nugroho AE, Morita H (2012) Chisomicines D and E, two new limonoids from Chisocheton ceramicus. Heterocycles 84:1265–1270
Supriatno S, Nurlelasari HT, Harneti D, Maharani R, Hidayat AT, Mayanti T, Supratman U, Azmi MN, Shiono Y (2018) A new limonoid from stem bark of Chisocheton pentandrus (Meliaceae). Nat Prod Res 25:1–7
Singh S, Garg HS, Khanna NM (1976) Dysobinin, a new tetranortriterpene from Dysoxylum binectariferum. Phytochemistry 15:2001–2002
Maneerat WS, Laphookhieo S, Koysomboon K, Chantrapromma K (2008) Antimalarial, antimycobacterial and cytotoxiclimonoids from Chisocheton siamensis. Phytomedicine 15:1130–1134
Haldar S, Kolet SP, Thulasiram HV (2013) Biocatalysis: fungi mediated novel and selective 12β-or 17β-hydroxylation on the basic limonoid skeleton. Green Chem 15:1311–1317
Yadav PA, Kumar CP, Siva B, Babu KS, Allanki AD, Sijwali PS, Jain N, Rao AV (2017) Synthesis and evaluation of anti-plasmodial and cytotoxic activities of epoxyazadiradione derivatives. Euro J of Med Chem 134:242–257
Pereira TB, Silva L, Amorim R, Melo M, Souza R, Eberlin M, Lima ES, Vasconcellos MC, Pohlit AM (2014) In vitro and in vivo anti-malarial activity of limonoids isolated from the residual seed biomass from Carapa guianensis (andiroba) oil production. Malar J 13:317
Examinati RRIN, Wulandari AP, Harneti D, Poniah A (2018) Cytotoxicity of aromatic compound from an endophytic fungus, Cladosporium sp EN-S01 Int J of Curr Pharm Res, 10, 10–12.
Funding
This investigation was financially supported by Universitas Padjadjaran, Indonesia (PPKI Grant, No: 1427/UN6.3.1/LT/2020 by US) by Airlangga University, Indonesia (PPKI Grant, No: 305/UN3.14/PT/2020 by MT) and Brawijaya University, Indonesia (PPKI Grant, No: 455.5/UN10.C10/PN/2020 by RR).
Author information
Authors and Affiliations
Contributions
NS, IR and SS: Isolation of limonoid from the n-hexane extract; AS, DH, RM, ATH, MT: Structural elucidation of isolated compounds; RR, DH: cytotoxic assay; NS, IR, US, YS: preparing and completing manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
There is so conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
1H-NMR Spectrum of 1 (500 MHz in CDCl3). Figure S2. 13C-NMR Spectrum of 1 (125 MHz in CDCl3). Figure S3. DEPT-135° Spectrum of 1 (135 MHz in CDCl3). Figure S4. HSQC Spectrum of 1. Figure S5. 1H-1H COSY Spectrum of 1. Figure S6. HMBC Spectrum of 1. Figure S7. 1H-1H-NOESY Spectrum of 1. Figure S8. HRTOFMS spectrum of 1. Figure S9. 1H-NMR Spectrum of 2 (500 MHz in CDCl3). Figure S10. 13C-NMR and DEPT-135° Spectrum of 2. Figure S11. HMQC Spectrum of 2. Figure S12. 1H-1H COSY Spectrum of 2. Figure S13. HMBC Spectrum of 2. Figure S14. 1H-1H-NOESY Spectra of 2. Figure S15. HRTOFMS spectrum of 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nurlelasari, Rahmayanti, I., Salam, S. et al. A new havanensin-type limonoid from Chisocheton macrophyllus. Appl Biol Chem 64, 35 (2021). https://doi.org/10.1186/s13765-021-00606-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13765-021-00606-5