
Abstract
Background
Activation of innate immunity is a first line of host defense during acute critical illness (ACI) that aims to contain injury and avoid tissue damages. Aberrant activation of innate immunity may also participate in the occurrence of organ failures during critical illness. This review aims to provide a narrative overview of recent advances in the field of innate immunity in critical illness, and to consider future potential therapeutic strategies.
Main text
Understanding the underlying biological concepts supporting therapeutic strategies modulating immune response is essential in decision-making. We will develop the multiple facets of innate immune response, especially its cellular aspects, and its interaction with other defense mechanisms. We will first describe the pathophysiological mechanisms of initiation of innate immune response and its implication during ACI. We will then develop the amplification of innate immunity mediated by multiple effectors. Our review will mainly focus on myeloid and lymphoid cellular effectors, the major actors involved in innate immune-mediated organ failure. We will third discuss the interaction and integration of innate immune response in a global view of host defense, thus considering interaction with non-immune cells through immunothrombosis, immunometabolism and long-term reprogramming via trained immunity. The last part of this review will focus on the specificities of the immune response in children and the older population.
Conclusions
Recent understanding of the innate immune response integrates immunity in a highly dynamic global vision of host response. A better knowledge of the implicated mechanisms and their tissue-compartmentalization allows to characterize the individual immune profile, and one day eventually, to develop individualized bench-to-bedside immunomodulation approaches as an adjuvant resuscitation strategy.
Similar content being viewed by others


Introduction
During acute critical illness (ACI), from sepsis to trauma, innate immunity is triggered instantaneously. Activation of innate immunity not only aims at eliminating pathogens, but also to avoid tissue damage and alleviate potentially harmful unregulated inflammation. While innate response represents a keystone of patients’ survival, aberrant activation may participate in the occurrence of organ failures.
This narrative review aims to provide a non-specialist audience with recent and relevant discoveries in the ACI-associated innate immune response field. Immunology is a rapidly growing scientific field, and advances in knowledge and technologies have fueled significant therapeutic progress in numerous medical areas, but not yet in intensive care. Nevertheless, intensivists now use diverse therapies to modulate the inflammatory response as part of clinical trials, and a large proportion of patients admitted in ICU undergo an immunomodulators or immunosuppressive treatments. This way, intensivists need to understand the underlying basic concepts supporting new personalized strategies.
Therefore, we will describe the ubiquitous and organ-specific pathophysiological mechanisms of activation of the innate immune response during ACI, with a focus on myeloid and lymphoid cellular effectors. Moreover, interaction and integration of innate immunity with other host defense systems will also be considered through immunothrombosis immunometabolism and trained immunity. The specificities of the immune response in children and elderly will also be discussed. In each section, we will first describe the known or putative molecular and cellular mechanisms involved in the activation of innate immunity.
Initiation of innate immunity
Initiation of the innate immune response requires the recognition of the aggression through evolutionarily conserved signals. During ACI, the accurate identification of these signals is essential to recognize the danger and stop its propagation.
PAMPs and DAMPs
Inflammation frequently results from the exposure of immune cells to pathogen-associated molecular patterns (PAMPs), which are conserved motifs expressed by microorganisms. Damage-associated molecular patterns (DAMPs), mainly represented by residues of necrotic cells produced in inflammatory processes, including non-infectious aggressions, as observed in trauma or burns [1], can alternatively trigger the immune response.
PAMPs are structurally diverse and exhibit a relative specificity to various pathogen groups (e.g. lipopolysaccharide (LPS) for gram-negative bacteria or mannane antigen for fungi). Thus, identifying conserved pathogenic motifs by innate immune cells enables the establishment of an initial level of specificity in response to the invading pathogen. Consequently, differential activation of the immune cells are observed depending on the nature of the recognized pathogen, e.g., cellular recognition of viral patterns results in local induction of type I interferon synthesis and, secondarily, activation, proliferation, and differentiation of cytotoxic T lymphocyte clones in the secondary lymphoid organs.
DAMPs are thus initiators of the so called ‘sterile inflammation’, and mechanistically lead to initiation of inflammatory cascades, in a similar -but not systematically redundant- ways to PAMPs [1, 2]. DAMPs encompass a wide variety of molecules, from both intracellular compartments to extracellular components. Prototypical DAMPs from intracellular origin are the DNA binding protein HMGB1, DNA, ATP, RNA, and mitochondrial components from stressed/damaged cells are known DAMPs able to promote inflammatory response once release out of their physiologic compartment. The term ‘alarmin’ is also sometimes employed to refer to these endogenous molecules from intracellular origin, and also include specific molecules like HMGB1, IL-33, S100, heat shock proteins that have similar roles [3]. Extracellular matrix components, such as hyaluronic acid or heparan sulfate are also classical DAMPs, released by matrix degradation during tissue injury. DAMPs are of high interest in ACI as they are associated with commonly seen pathophysiological mechanisms: ischemia–reperfusion mechanisms, tissue injury (trauma). Moreover, link with DAMPs released in critically ill patients have been associated with poor outcomes [4].
PRMs—pentraxins
Humoral part of innate immunity includes soluble recognition molecules (PRMs) that recognize PAMPs and DAMPs.
PRMs notably encompass the pentraxin family, consisting of proteins that possess a shared domain and are constructed from monomers organized into multimeric structures with a disc-like form. A defining feature of this family is a conserved sequence of 205 amino acids located at the C-terminal, referred to as the pentraxin domain. Based on the overall length of their protein sequences, the pentraxin family can be divided into two subgroups: short and long pentraxins. Short pentraxins include proteins like C-reactive protein (CRP, also known as pentraxin 1 or PTX1) and serum amyloid P component (SAP, also known as pentraxin 2 or PTX2), while long pentraxins encompass proteins such as neuronal pentraxin 1 (NPTX1), neuronal pentraxin 2 (NPTX2), neuronal pentraxin receptor (NPTXR), pentraxin 3 (PTX3), and pentraxin 4 (PTX4) [5, 6]. Numerous studies have shed light on the specific roles played by certain members of the pentraxin family. Notably, CRP and SAP have been recognized for their regulatory functions within the human immune system including defending against pathogens, linked to their capability to attach to a variety of bacteria, fungi, and viruses, thereby bolstering the innate immune responses against these pathogens [5, 7,8,9,10]. Moreover, pentraxins bind to phospholipids and certain nuclear ribonucleoproteins within apoptotic cells, facilitating their non-inflammatory clearance. C-reactive protein, SAP, and PTX3 engage with diverse complement molecules, thereby amplifying their recognition capabilities, and facilitate the phagocytosis of microbes and apoptotic cells through interactions with FcγR [11, 12]. The two-sides of the discoid structure of pentraxins seem to exhibit complementary features of the aforementioned functions. This is notably illustrated in CRP, with one side being involved in the activation of the classical complement pathway, thereby facilitating phagocytosis, whiles the other side interacts with bacterial cell walls phosphorylcholine, thereby leading to pathogen clearance [13, 14].
PRRs
PAMPs and DAMPs are recognized by tissue-resident immune cells (mast cells, macrophages and tissue dendritic cells -tDCs-) but also by epithelial and endothelial cells, expressing pattern recognition receptors (PRRs). Once activated, these cells release inflammatory mediators (pro-inflammatory cytokines, lipid derivatives…), activating endothelial cells and initiating the vascular phase of the inflammatory response.
PRRs can interact with numerous antigenic patterns, including LPS, peptidoglycan, viral RNA or bacterial DNA. Their localization (on cellular or endosomal membranes, in the cytosol or secreted [15]) generally fits with the cellular compartment in which the recognized pathogens are found.
While PRRs play a key role in initiating and amplifying the immune response, which is essential for the early recognition and control of aggression, they can also turn into a dysregulated response, subsequently leading to organ failure through cellular and tissue damage.
Membrane PRRs, known as Toll-like receptors (TLRs) induce cell activation via NF-kB-dependent signaling cascades (modulation of genes coding for pro-inflammatory molecules or co-stimulatory molecules essential for activating the adaptive response). These receptors have various ligands, including peptides, carbohydrates, lipids, DNA, and RNA. Several types of TLRs are observed in primary effectors of the immune response like neutrophils, including TLR1, − 2, − 3, − 4, − 5, − 6, − 8, and − 10, each of them recognizing various ligands. The engagement of these TLRs contributes to the initiation of the innate immune response through various mechanisms such as the TRIF pathways, involved in the production of type I interferon and pro-inflammatory mediators following the activation of TLR3 and 4 [16]. This priming of innate response secondarily stimulates the secretion of cytokines by innate immune cells and enhances phagocytosis [17,18,19].
C-type lectin receptors (CLRs) like Dectin-1 are major PRRs meaning they induce the phagocytosis of recognized pathogens. Dectin-1 is notably involved in the internalization and killing of fungi, through fungal β-glucans antigens recognition [20, 21].
Nucleotide-binding Oligomerization Domain (NOD) 1 and NOD2 are other PRRs localized in the cytosol, interacting with peptidoglycan-related molecules. They belong to the larger family of NOD-like receptors (NLRs). The stimulation of NOD2 by its ligand triggers the secretion of IL-8 in neutrophils. Activation of cytosolic NLRs also leads to cell activation (synthesis of the pro-inflammatory cytokine IL-1) [22, 23].
Signal integration & amplification during innate & inflammatory response
This wide range of PRRs allows specific identification of the nature of the threat, and, at the cellular level, multiple signals from PAMPs and DAMPs are integrated and regulated to produce tightly modulated effector response [24,25,26]. These varied pathways contribute to the diversity of observed profiles based on the origin of the inflammatory response. Recent research highlights that patient phenotypes predominantly rely on the source of the initial aggression [27]. Interestingly, beyond recognition by PRR of foreign versus endogenous ligands, many immune & barrier cells can also sense a harmful / stressful environment both at extra and intracellular levels. This is notably done by the cytosolic multiproteic complexes called ‘inflammasomes’ and especially the NLRP3 inflammasome [28]. Once assembled and activated, NLRP3 inflammasome lead to caspase 1 activation that can produce mature IL-1β, but also leading to necroptosis, an inflammatory death cell [29]. Thus, important cellular perturbation resulting from an initial insult can lead to inflammatory death, locally perpetuating inflammatory signal. Given that other inflammatory cell deaths have been discovered during the last decades, it has been proposed that inflammation and inflammatory cell death could self-perpetuate in organs after initial insult, thus contributing to organ failure [30].
Effectors of innate immunity
Recognition of danger signals induces rapid mobilization of immune cells from the bone marrow to the injury site. Immune activation aims simultaneously to limit tissue invasion and initiates the resolution of inflammation (Fig. 1).

Innate immune response. Innate immune response is characterized by both proinflammatory (in red, top of the figure) and anti-inflammatory (in green, bottom of the figure) responses. Pro-inflammtory response can results in cellular damages forming DAMPs, that themselves trigger inflammation pathways. Anti-inflammatory response can induce immunoparalysis, resulting in secondary infections. The balance of this host response, its duration and its intensity are dependent of multiple factors: type of aggression on one hand, host factors (genetic, immune state, medical history, age …) on the other hand. C1 complement 1, DAMPs danger associated molecular patterns, DHA docosahexaenoic acid, EPA eicosapentaenoic acid, IL interleukin, NETosis release of NETs, Neutrophil Extracellular Traps, PAMPs pathogen associated molecular patterns, PLA2 phospholipase 2, ROS reactive oxygen species, TLR toll like receptor, TNF tumor necrosis factor
This section focuses on the understanding of myeloid and lymphoid activation and dysfunction, and their potential consequences for clinicians through innovative therapy targeting innate immune cells. A recently published review thoroughly describes the humoral part of innate immunity, and will thus not be reviewed here [31].
Myeloid cells
From host defense to tissue damage
Myeloid cells, including monocytes, macrophages, neutrophils, and DCs, are the first cells activated in early response to a danger signal. Neutrophils are sensitive to different inflammatory, infectious or physical signals which induce their release from bone marrow, intravascular and transendothelial migration to the aggression site [32, 33]. Phagocytosis was long considered as the primary function of macrophages and neutrophils on the injury site, especially considering infections [34]. Phagocytosis occurs through different mechanisms. One is oxygen-dependent and mediated by the reactive oxygen species (ROS). ROS are highly toxic, and capable of bacterial destruction but also participate to tissue injuries [35]. High levels of ROS are associated with early death and post-aggressive immunodepression, and secondary infections. Mitochondria are the main source of ROS production, and therefore appear as key components of the immune regulation occurring in critically patients. It seems noteworthy to mention that mitochondrial ROS not only participate to direct bacterial killing, but upregulate the synthesis of pro-inflammatory mediators as well, through activation of the NLRP3 inflammasome [36]. Unbalanced ROS production in myeloid cells may directly inflict local damages to mitochondrial proteins and DNA, thereby leading to significant impairment of their function during the immune response [37].
This example illustrates the complex functional ambiguity of myeloid cells during ACI: while their role in host defense is essential for survival, their activation can also become aberrant and participate in organ dysfunctions [38]. Thus, overstimulation of the bone marrow also triggers the release of immature monocytes and neutrophils, called myeloid-derived suppressive cells (MDSCs). Circulating immature granulocytes present altered effector capacity (i.e. pathogen recognition, reduced phagocytosis capacities,) and their abundance is associated with early mortality and secondary immunosuppression [39,40,41]. MDSCs also participate to tissue hypoperfusion through microvessels obstruction due to their immature rheological properties [42]. Monocytic MDSCs are characterized by a decreased surface-expression of human leucocyte antigen HLA-DR, commonly associated with nosocomial infections and secondary-immunosuppressive state [43].
Lipid mediators of innate immunity
In response to primary inflammatory mediators (IL-1, Tumor Necrosis Factor -TNF-,) myeloid cells synthesize phospholipase A2 (PLA2) that transforms membrane phospholipids in arachidonic acid. Released free arachidonic acid can be further metabolized (i) by cyclooxygenase (COX) 1 and 2 to prostaglandins (PG) and thromboxanes (TXA), and (ii) by lipoxygenase (LO) to leukotrienes (LK) and lipoxins [44]. These molecules are named eicosanoids. They are rapidly metabolized, resulting primarily in a local action due to their short lifespan [45].
Eicosanoids are critical actors in the regulation of inflammation [28] during ACI, by regulating vasodilatation-vasoconstriction balance, vascular leakage, and platelet activation. One example in ACI is the elevation of circulating Platelet-activating factor (PAF), during critical phase of dengue hemorrhagic fever that potentially promotes major capillary leak syndromes [46]. Eicosanoids and other lipids are also implicated in the resolution of injury through anti-inflammatory effects [47]. Lipoxins can inhibit macrophages and neutrophils recruitment. These specialized pro-resolving mediators (SPMs) include other lipid molecules like omega-3 derived fatty acids that have been isolated in inflammatory exudates [48]. SPMs also participate to bacterial clearance and efferocytosis. Because of their immunomodulation properties, nutritional supplementation of omega-3 fatty acids was proposed in septic patients, but did not improve mortality [49].
Inhibition of the formation of eicosanoids by aspirin or non-steroidal anti-inflammatory drugs is a classical therapy to prevail fever or pain. Nevertheless, eicosanoids synthesis is very dynamic and compartmentalized, with tight organ-specific regulation. This complexity may explain the failure of clinical trial testing inhibition of COX2 by ibuprofen during septic shock [50]. A more extensive understanding of the activated pathways and the identification of the side products of biosynthesis using new tools like mass-spectrometry lipidomic profiling is needed [51, 52].
Interplay between macrophages and neutrophils
Recent advances in technologies exploring immunity helps uncovering novel functions of myeloid cells. Various subpopulations of macrophages have been identified across different tissues and exhibit various functions beyond phagocytosis [38]. Ischemia–reperfusion, which is observed in a wide range of ACI, is characterized by an initial restriction of oxygen supply to an organ before perfusion is restored. Hypoxia induces endothelial dysfunction [53] and results in the activation of various cellular cell death programs including NETosis (release of Neutrophil Extracellular Traps), apoptosis and autophagy [54]. Like cytokine storms, excessive release of NETs and apoptotic cells may exacerbate the inflammatory state during sterile aggressions, thus participating in the development of acute respiratory syndrome (ARDS) as observed in trauma patients [55]. Surprisingly, experimental studies have shown that increased neutrophil lifespan is associated with a deleterious impact [56]. This could be explained by the reduced clearance of NETs and apoptotic cells by macrophages, called efferocytosis [57]. Cytoskeletal modifications of macrophages are required for this function. Experimental studies have identified the inhibition of AMP-activated protein kinase (AMPK) as a significant efferocytosis contributor [58]. Inhibition of AMPK activity is observed during ARDS [59]. Restoration of AMPK activity, and thus macrophage function, represents a promising target for reducing lung inflammation [57].
Interaction with adaptive immunity
Overall, the immune response is highly dynamic and myeloid cells interact with other cells. Neutrophils can directly activate DCs through DC-SIGN receptors expressed on the surface of immature DCs and Mac-1, and this interaction is essential for both cellular functions [60]. DCs are responsible for the initiation of antigen-specific immune responses. The direct interaction with neutrophils orientates the polarization of lymphocytes to a Th1 phenotype [61], but also requires a favorable microenvironment including the presence of TNFα. DCs could thus play a major role in immune regulation, and represent a potential effector for the development of therapeutic vaccines [62]. Myeloid cells are also able to stimulate or inhibit B-lymphocytes in the lymphoid organs, depending on their microenvironment.
Innate lymphoid cells and innate T cells
Immune cells of lymphoid origin differ from myeloid cells during their ontogeny in the bone marrow. The common lymphoid progenitor (CLP) first emerges from hematopoietic stem cells (HSCs). Then, this CLP can remain in the bone marrow and engage in the innate lymphoid lineage, or engage in T cell fate, by reaching the thymus and pursuing their dedicated specific T cell maturation.
Innate lymphoid cells
ILCs are a peculiar and heterogenous population of immune cells. However, they do not express a diversified antigen receptor, therefor excluding them from T and B lymphocytes families, and can rapidly provide their effector function. Thus, they are considered to be part of the innate immune system [63]. From a functional point of view, ILCs are often considered as the innate counterpart of conventional T cells and have emerged as key players in the early orchestration of the immune response, particularly at the barrier sites (lung, skin, gut) [64]. Different subsets have been described, schematically mirroring those known for T cells: some subsets are known to have “helper” properties, -in a similar way to CD4+ T helpers-, by producing cytokines according to the type of aggression (termed ILC1, 2 and 3). Similarly, the so-called “Natural-Killers” cells are classified as ILC and mirror the cytotoxic CD8+ T cells.
ILCs are usually not the first responders among the innate immune arm but are instructed by signals (cytokines) provided by the cells that contact pathogens/aggressors. However, their strategic position at barrier sites allows them to be rapidly informed of local modification in the environment and potential threats. Indeed, ILCs are mostly tissue-resident cells with the notable exception of circulating NK cells that can represent up to 15% of circulating lymphocytes. Tenrichment in different subsets of ILCs varies according to the tissue considered [64]. Besides aggression, ILCs also contribute locally to the maintenance of homeostasis, especially in the gut, through the control of epithelial integrity and interaction with the microbiome [64, 65]. Last, ILCs also shape the adaptive response, either through soluble mediators acting on adaptive cells or by direct cellular interaction with adaptive cells [66].
In vivo studies in mice using viral, bacterial and fungal models of lung, skin, and gut infections have shown the involvement of ILCs in the control and resolution of infection [64, 67, 68]. Protection is thus conferred by secondary signals released by ILCs to induce appropriate immune response: IL-17 and IL-22 produced in response to extra-cellular pathogens induce local production of anti-microbial peptides and recruitment of neutrophils; IFNγ produced during infection by intra-cellular pathogens induces the recruitment of phagocytes etc. NK cells also have cytotoxic properties, that confer protection notably against intra-cellular infections (but also against tumoral cells). Regarding pathogens experimentally evaluated in this field, many are potentially relevant in the setting of ACI: influenza, S. pneumoniae, K. pneumoniae, C. difficile, C. albicans. It is also noteworthy that evidence exists for the involvement of ILCs in tissue repair after acute injury, notably ILC3 and ILC2, for example after influenza pneumonia [65].
However, to date in human, data from patients during disease are scarce, especially for ILC1, 2 and 3 subpopulations, recently discovered and technically more complex to analyze. Studies, due to obvious ethical and technical limitations, are mostly descriptive and limited to chronic inflammatory condition (i.e. asthma, psoriasis, [69, 70]), apart from the recent publication of ILCs alteration in function and frequency in peripheral blood during COVID-19 [71]. Data accumulated regarding NK cells in human, however, are more extensive, revealing alteration in blood frequency and function of NK cells during sepsis, but associations with outcome are controversial [72]. It is reasonable to hypothesize that NK cells implication in sepsis might vary according to the considered stage of the disease (i.e., early course of sepsis versus post-aggressive immunesuppressive phase).
At the end, the discovery of these cells and data accumulating regarding their implication in numerous physiological and pathological settings are redefining our way of considering the innate immune response and could be a game-changer in a near future.
Innate T cells
The term “innate T cells” (ITCs), is used to describe subpopulations of T cells that are endowed with specific properties and functions, differing from conventional adaptive T cells, and ontogenetically, they differentiate from conventional arm during thymic maturation [73]
ITCs have unique and complementary properties compared to ILCs and conventional adaptive T cells, and they are thus strategically poised at the interface between adaptive and innate immunity, which they can both modulate and shape according to the type of threats they face. Moreover, ITCs are enriched at barrier sites where they can exert their versatile functions ranging from initiation and amplification of the immune response to tissue repair [74]. All these properties make ITCs potentially attractive targets in the immunopathology of ACI. The recent development of specific tools for their detection led to a growing body of evidence of their implication in ACI, notably Mucosal-Associated Invariant T (MAIT) cells, as exemplified by the COVID-19 pandemic [75,76,77].
Innate T cells are also described as “preset T cells”, capable of rapid activation, similar to innate immune cells [78]. Moreover, unlike conventional adaptive T cells, they do not need to go through a clonal expansion phase after being stimulated. Two main lineages of ITCs are described: invariant Natural Killer T (iNKT) cells and MAIT cells. A third population can be added to the “innate T cells” subgroup, comprising some T cells harboring a TCR built with specific γ and δ chains (mainly, in humans those harboring Vγ9 Vδ2 chains) [79].
Through their TCR, MAIT and iNKT cells recognize small non-peptidic antigens presented by non-polymorphic MHC-related molecules: thus, ITCs harbor TCR and recognize antigens invisible to conventional T cells -that recognize small peptides. Moreover, ITCs can be activated through TCR-independent stimulation, notably via cytokines.
Upon stimulation, Innate T cells can exert multiple immune functions, from cytotoxicity to tissue repair [74]. Akin to ILC and conventional T cells, some effector functions of ITCs can also be considered as “helpers”, depending on the cytokines they produce.
A substantial body of evidence has demonstrated the implication of ITCs in various experimental models of ACI. Relevant models of bacterial and viral pneumonias have demonstrated the beneficial role of ITCs during infection through the orchestration of immune response and tissue repair [80, 81]. However, how these cells could be implicated in dysregulated host response as observed in ARDS and other ACI remains largely unknown and justify in-depth analysis of these cells.
In clinical settings, the implication of ITCs has been observed in patients with sepsis, severe influenza, and severe COVID-19 patients [75,76,77, 82, 83]. Both iNKT and MAIT cells frequencies in the blood were drastically reduced in these conditions, while presenting activation markers. Association with patients’ outcomes has also been explored, highlighting a correlation between MAIT cell persistent deficiency and the frequency of nosocomial infections [82]. Moreover, in severe COVID-19, MAIT cell activation was associated with poor outcome in three studies while being associated with better outcome in another study [75,76,77]. These discrepancies, possibly due to differences in patients’ severity suggest that MAIT cells might have dual and opposite functions, according to environmental cues [84].
ITCs are already considered as attractive targets for immune intervention, notably because of their vast array of functions and their specificity for public antigens presented by conserved and non-polymorphic molecules. Thus, several clinical trials are already ongoing in other medical fields (especially oncology) [85].
Immunothrombosis
In response to aggression, vascular cells, including innate immune cells, platelets, and endothelial cells are activated and trigger innate immunity and coagulation pathways. The crosstalk between immunity and coagulation was thus recently defined by the term “immunothrombosis” [86]. It refers to an innate intravascular immune mechanism that recognizes and contains the injury through the activation of innate immunity and coagulation, leading to the formation of “protective” thrombi in microvessels [86]. Unbalanced and dysregulated immunothrombosis results in inadequate host response during infectious diseases and sterile ACI like myocardial infarction, stroke or trauma.
Neutrophils recruited to the site of infection in response to chemoattractant molecules like CXCL2 release the content of their granules and ROS. Platelets are activated through the binding of glycoprotein (Gp) Ib-V-IX to Willebrand Factor [86]. They release the content of their granules, including RANTES, PF4 and CD40L [87] and interact with myeloid cells via direct binding to different receptors like Mac-1 (Macrophage 1 antigen) expressed on myeloid cells [88]. Monocytes, platelets and neutrophils also release microvesicles (MVs) [89, 90]. Tissue factor (TF) expression on the surface of the activated cells, MVs, NETs, and some PAMPS and DAMPS contribute to the coagulation pathway activation to contain the zone of injury. The interactions between coagulation and immunity thus represent a first line of defense. During ACI, the severity of the injury and the inadequate host response with deregulated immunothrombosis promote a hyper-inflammatory and hyper-oxidative state [91]. Subsequently, the previously described impaired myeloid functions, including neutrophil CCR2 expression, lead to their accumulation in organs, thus contributing to multiple organ failure [92]. The systemic inflammation also induces an exaggerated and uncontrolled NETosis, participating in capillary occlusions. Endothelial dysfunction promotes leukocyte and platelet adhesion, coagulation activation, and fibrinolysis inhibition. Ultimately, uncontrolled thrombin formation (excessive activated thrombin FIIa formation) leads to disseminated microthrombi, capillary occlusions, and subsequent impaired organ perfusion. The main clinical issue revealing the deregulated immunothrombosis is disseminated intravascular coagulation observed during different ACI as diverse as trauma, septic shock or obstetrical diseases. Exaggerated immunothrombosis can also be a compartmentalized phenomenon, for example in Sars-CoV2-infected lungs [93,94,95] and also in non-COVID ARDS [96] or after surgery [97] (Fig. 2).

Immunothrombosis in acute critical illness. Immunothrombosis is a local defense mechanism that can be dysregulated during ACI, inducing an hyper-inflammatory and hyper-oxidative state. Impaired immunothrombosis is notably mediated by neutrophils and endothelial dysfunction, activated leukocytes and platelets. Dysregulated immunothrombosis results in microthrombi formation, responsible of various organ dysfunction during ACI. MVs microvesicles, NETs neutrophil extracellular traps, ROS reactive oxygen species, TF tissue factor
Numerous randomized clinical trials have failed to demonstrate the benefits of anticoagulation and anti-inflammatory therapies during ACI, especially in septic shock. The better comprehension of the crosstalk between innate immune cells and non-immune effectors offers new hopes for developing targeted therapeutics, i.e. by targetting NETs. The pathogenicity of NETs is no well demonstrated in animal models. Besides, NETs are implicated in tissue damages in a relevant murine model of lesional pulmonary oedema [55]. The administration of anti-histone antibodies prevented the extension of pulmonary lesions. In another model of acute lung injury in mice, DNase and inhibitor of neutrophil elastase also attenuated lung injury [98].
Trained immunity
Trained immunity defines the innate immunological memory orchestrated by epigenetic reprogramming. This concept encompasses changes in gene expression and cellular physiology without permanent genetic changes, such as epigenetic modifications. These chemical changes, including DNA methylation, histone modification, and RNA-associated silencing can affect gene expression without altering the underlying genome. These modifications can have a wide range of effects on gene expression, including genes activation or repression, thus modulating the immune response.
Until recently, it was commonly accepted that training and memory abilities were the hallmarks of adaptive immunity. However, several discoveries have challenged this paradigm.
Indeed, it has been shown that exposure to a pathogen could modulate the long-term immune response via the functional reprogramming of innate immune cells. This hypothesis was based on results showing that BCG vaccination surprisingly led to an increased long-term response to β-1,3-D-glucan in the wall of Candida albicans [99]. This phenomenon was mediated by specific and persistent changes in histone acetylation and methylation [100,101,102,103]. These findings allowed the identification of acquired and persistent immune response alteration, called innate immune memory, consisting either in an increased response to restimulation, called trained immunity, or a reduced immune response, called immune tolerance [104]. Finally, it should be noted that this phenomenon may directly concern the cells of innate immunity and their precursors, which can potentially transmit these alterations in immune memory [105].
Numerous inducers of immune training have been described based on experimental data. These different factors are thus likely to modify the phenotype of the response to aggression in case of prior exposure in ACI. Thus, we can mention Candida albicans and BCG as previously described, but also Mycobacterium tuberculosis [106], viral agents like HIV and HBV [107, 108], Plasmodium falciparum [109] or the diphtheria/tetanus/poliomyelitis/pertussis combined vaccine [110]. It is also interesting to note the potential influence of non-infectious factors on immune training, like diet [111], physical exercise [112], or circadian rhythm [113].
The pathways by which these different factors influence the long-term immune response have been widely studied. The activation of several metabolic pathways, including glycolysis, oxidative phosphorylation and lipid metabolism, plays a significant role [114,115,116]. These mechanisms generate numerous mediators which play a role in chromatin modifications involved in innate immune memory [116, 117].
From a clinical point of view, the innate immune training seems to be associated with a protective effect against specific pathogens. This was particularly illustrated during the COVID-19 pandemic. Indeed, several authors reported the potential protective role of BCG vaccination against SARS-CoV-2 infection [118, 119]. However, these beneficial effects seem to be counterbalanced by the existence of deleterious effects linked to innate immune memory, in the fields of chronic inflammatory diseases [120], allergology [121] or organ transplantation rejections [122].
Age related features of innate immunity
Pediatric specificities
Infants’ immune system is still in development, and presents specific characteristics including impaired PAMPs and DAMPs recognition.
Two intrinsically linked mechanisms will be considered when discussing innate immunity in the intensive care units (ICU). The first question is the reason why some children have severe infectious diseases, with the hypothesis that such life-threatening disorders could be due to a pre-existing immune deficiency. The second is the consequences of ACI on the innate immune system.
Exploring how innate immunity deficiencies can lead to life-threatening acute illnesses in children permitted the identification of many immune deficiencies [123]. In young children, given the absence of a fully mature adaptive immunity, the innate immune system plays a crucial role in fending off infections. Indeed, rare deficiencies in the complement system have been shown to lead to disseminated meningococcal infection and/or meningitis [124]. In turn, such severe infection leads to uncontrolled inflammation and organ injuries. Similarly, in children with severe or critical COVID-19 pneumonia, deficiencies in the critical mediators of early antiviral defenses type I IFN have been shown to underlie more than 10% of cases [125, 126]. Moreover, in some children, inborn errors of OAS–RNase L can, following a trigger by SARS-CoV-2, unleash the production of MAVS-mediated inflammatory cytokines by mononuclear phagocytes, thus leading to a Kawasaki-like disease, called multi-inflammatory syndrome – children [127, 128].
ACI-associated innate immune disruption in children is hardly quantifiable as intimately intricated with the cause of severe disease. Nevertheless, in non-infectious settings in patients with acute organ damage in the ICU, several consequences on the innate immune system have been observed like immune paralysis [129] and reduction in key immune molecules such as HLA-DR, therefore facilitating the development of nosocomial infections [130].
Newborn children are a specific group as their immunity is developing and changing rapidly [131]. They are also highly susceptible to infections, for reasons that are not well understood [132].
Overall, identifying such immune deficiencies could guide the clinical strategy in children with ACI. It not only represents a challenge because of the consequences of inflammation or organ injuries, but more importantly because these deficiencies can be the potential underlying cause of the severe disease.
Immunosenescence
Immunoscenescence refers to the progressive decline of immune functions with aging. The two pillars of this immune aging are a reduced capacity of response to pathogens and antigens -such as in vaccinations-, and a chronic low-grade systemic inflammation without evident trigger, called ‘inflammaging’ [133] (Fig. 3). The older population being heterogeneous, the level of immunosenescence and its consequences are highly variable between individuals of the same age.

Innate immunity in aging patients. Balance between inflammatory and anti-inflammatory is highly modified in aging. The two main altered mechanisms are: a reduced capacity of response to injury by pro-inflammatory senescent cells (right) and a chronic systemic inflammation called inflammaging (left). CD cluster of differentiation, DNA desoxyribonucleic, IL interleukin, LT lymphocyte T, NK natural killer, ROS reactive oxygen species, SASP senescence-associated secretory phenotype, Th T helper, TLR toll like receptor, TNF tumor necrosis factor
Impact of aging on innate immune cells
Virtually all innate immune cells are affected with aging. First, HSC progenitors have a decreased functionality with altered expression and inappropriate activation of PRRs, and the distribution between lymphoid and myeloid progenitors is imbalanced, in favor of myeloid progenitors [133]. Regarding mature functional cells, the absolute number of innate immune cells shows little variations with age, but profound functional changes occur. Neutrophil have an increased clearance rate and their phagocytosis and chemotaxis capacities are reduced (after LPS stimulation) [134,135,136]. Monocytes display altered phenotypes along with functional changes: impaired phagocytosis, and impaired TLR1 and 2 pathways [137,138,139,140]. Macrophages display different alteration profile with ageing, according to their tissue residency and ontogeny [141, 142]. Their phagocytic capacities -including efferocytosis- appear impaired, but cytokine production might be increased in some conditions/subsets (thereby contributing to inflammaging), and reduced in others [143, 144]. DCs have also altered phagocytic and antigen presentation, and upon TLR stimulation, pro-inflammatory cytokine secretion is reduced, leading to impaired T cell priming and defective CD4 + T cell polarization [145].
Innate cells from lymphoid origin are also altered with ageing, though data are relatively scarce. Among ILCs, group 2 ILC from aging mice have been reported to have lower response during influenza infection [146]. Regarding ITCs, blood MAIT decreased in the blood during adulthood, potentially associated with functional alteration [147]. Similarly, blood levels of γδ T cells harboring Vδ2 chain decline with age andtheir phenotype is altered [148], but the clinical consequences of these observed phenomenon are still unknown.
Inflammaging
Modification of innate cells activation generates a dysregulated level of inflammation which participates in inflammaging characterized by a chronic increase in serum levels of pro-inflammatory cytokines such as IL-6, TNF-α, IFN-a and IL1-β [149]. Although still incompletely understood, this phenomenon results from several mechanisms: (i) inappropriate activation of PRRs by self-debris from defective autophagy, mitophagy and ubiquitin/proteasome system [150, 151], (ii) dysbiosis [152], (iii) mitochondrial dysfunction [153,154,155], (iv) cellular senescence with their “senescence-associated secretory phenotype” (SASP) and tissular aging [153, 156], (v) endoplasmic reticulum stress [157], (vi) and DNA damage [153]. Consequences include oxidative stress, tissular lesions, alteration of metabolism and endocrine system and accumulation of MDSCs. Inflammaging is associated with worse outcomes in older patients, with increased mortality, comorbidities, sarcopenia and frailty [158]. Inflammaging is not only a consequence of immunosenescence, as it plays a role in maintaining and aggravating immune cell senescence through adaptive immune cell exhaustion [159].
Inflammaging and altered innate immune cell function result in increased susceptibility to bacterial and viral infections. As a consequence, the incidence and severity of sepsis increases in the older population [160, 161]. Furthermore, some sepsis survivors have persistent inflammation reminiscent of accelerated immune aging, feeding a vicious cycle [162, 163].
No specific studies on therapies targeting the innate compartment during sepsis have been performed in the older population. Several have been explored, including immunoglobulins (to neutralize endotoxins and improve monocyte/macrophage phagocytic ability), IFN-g and GM-CSF (to enhance neutrophils and monocytes/macrophages phagocytosis, and cytokine release). To date, none have shown any efficacity on mortality [164]. Boosting immune system could be even deleterious [165, 166], and other clinical trials exploring the approach of immune boosting are underway.
Immune response: behind the phenotype
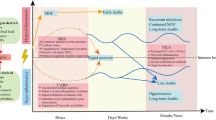
Heterogeneity of the immune response remains as an important limiting factor for the development of targeted treatments in the field of acute inflammation. Recent data underline the difficulties of understanding the phenotypic specificities of each patient. Numerous studies have shown that patients who appear to be similar at bedside, due in part to similar sources of infection or type of aggression, may in fact differ in terms of the inflammatory response revealed by next-generation approaches [167]. The ability to group patient according to shared common pathophysiological processes could help to refine prognosis performance, and ultimately identify patients that could benefit from targeted therapies. Such categorization beyond clinical phenotypes is called ‘endotypes’ [168, 169]. The benefits of such an approach in patients characterization were notably highlighted by Antcliffe, et al., who showed that a transcriptomic analysis could distinguish subgroups of patients according to their degree of response to corticosteroid therapy during sepsis [170]. One important challenge in the field of critical illness remains to clearly define the scope of application of these new approaches, in order to better personalize the management of patients. Specifically, given importance of dysregulation of immune response in ACI, researchers should focus on defining immune endotypes from translational immunology discoveries in ACI. However, one of the major drawbacks for these potential therapeutic avenues is the lack of consideration of spatial dynamics and spatial specificities of innate immune response during ACI. Spatial considerations of immune response refer to the concept of compartmentalization of immune response: immune response to aggression, at a considered time-point, might differ in intensity and modality across anatomical sites of the body. This especially relevant in many situations in critical care where initial insult is frequently located at a given site, with subsequent systemic diffusion of inflammatory response. Thus, we and others demonstrated that during pneumonia-driven ARDS, inflammatory response -explored through cytokines concentrations- was largely compartmentalized to the lung [171, 172]. Moreover, beyond consideration of soluble mediators, cellular actors of innate immune response are particularly subject to phenotypic and functional alteration according to the organ considered (example: blood monocyte versus various multiple macrophages subsets). Consequently, sampling blood to monitor immune response might not be adequate to infer immune status of other distant anatomical sites [173].
Conclusion
The ACI-associated systemic inflammatory response reflects the complex and highly dynamic host response mediated by the innate immune system. The overactivation of the immune response, partly responsible for critical illness, notably results from the synergy between the various mechanisms involved in the inflammatory response leading to its disproportionate amplification [174, 175].
In recent decades, milestones have been reached in understanding this response and its consequences in acute and chronic organ dysfunction. Some of these knowledge have already been translated into clinical trials targeting specific host pathways [176, 177]. However, the diversity of the underlying diseases and the highly variable host response makes the bridge from bench to bedside highly hazardous. Thus, many efforts have still to be made before considering future routine implementation of targeted host modulation strategies in critical care medicine. It is assumable that building new tools allowing precise real-time endotyping of the individual innate response during ACI are a preliminary necessary step before unleashing personalized immunomodulation of the innate response as part of resuscitation strategies.
Availability of data and material
Not applicable.
Abbreviations
- ACI:
-
Acute critical illness
- AMPK:
-
AMP-activated protein kinase
- ARDS:
-
Acute respiratory distress syndrome
- BCR:
-
B cell receptors
- C1:
-
Complement 1
- CD:
-
Cluster of differentiation
- COX:
-
Cyclooxygenase
- CLP:
-
Common lymphoid progenitor
- CRP:
-
C-reactive procedure
- DAMPs:
-
Danger-associated molecular patterns
- DCs:
-
Dendritic cells
- DHA:
-
Docosahexaenoic acid
- DNA:
-
Desoxyribonucleic
- EPA:
-
Eicosapentaenoic acid
- HLA:
-
Human leucocyte antigen
- HSCs:
-
Hematopoietic stem cells
- ICU:
-
Intensive care unit
- ILC:
-
Innate lymphoid cells
- IFN:
-
Interferon
- IL:
-
Interleukin
- ITCs:
-
Innate T cells
- LK:
-
Leukotriene
- LTCI:
-
Long-term cognitive impairment
- LPS:
-
Lipopolysaccharide
- LT:
-
Lymphocyte T
- MAIT:
-
Mucosal-associated invariant T
- MDSC:
-
Myeloid-derived suppressive cells
- MVs:
-
Microvesicles
- NETs:
-
Neutrophil extracellular traps
- NK:
-
Natural killer
- NLRs:
-
NOD-like receptors
- NOD:
-
Nucleotide-binding oligomerization domain
- PAF:
-
Platelet-activating factor
- PAMPs:
-
Pathogen-associated molecular patterns
- PLA2:
-
Phospholipase 2
- PG:
-
Prostaglandin
- PRMs:
-
Recognition molecules
- PRRs:
-
Pattern recognition receptors
- PTX:
-
Pentraxin
- ROS:
-
Reactive oxygen species
- SASP:
-
Senescence-associated secretory phenotype
- SPM:
-
Specialized pro-resolving mediators
- TCR:
-
T cell receptors
- Th:
-
T helper
- TF:
-
Tissue factor
- TLC:
-
Total lung capacity
- TLR:
-
Toll like receptor
- TNF:
-
Tumor necrosis factor
- TXA:
-
Thromboxanes (TXA)
References
Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37.
Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112.
Yang D, Han Z, Oppenheim JJ. Alarmins and immunity. Immunol Rev. 2017;280:41–56.
Timmermans K, Kox M, Scheffer GJ, Pickkers P. Danger in the intensive care unit: damps in critically Ill patients. Shock. 2016;45:108–16.
Pepys MB. The pentraxins 1975–2018: serendipity diagnostics and drugs. Front Immunol. 2018;9:2382.
Gonzalez OA, Kirakodu S, Novak MJ, Stromberg AJ, Orraca L, Gonzalez-Martinez J, et al. Comparative analysis of microbial sensing molecules in mucosal tissues with aging. Immunobiology. 2018;223:279–87.
Thompson D, Pepys MB, Tickle I, Wood S. The structures of crystalline complexes of human serum amyloid P component with its carbohydrate ligand, the cyclic pyruvate acetal of galactose. J Mol Biol. 2002;320:1081–6.
Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. 2015;373:1106–14.
Richards DB, Cookson LM, Barton SV, Liefaard L, Lane T, Hutt DF, et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci Transl Med. 2018. https://doi.org/10.1126/scitranslmed.aan3128.
Doni A, Parente R, Laface I, Magrini E, Cunha C, Colombo FS, et al. Serum amyloid P component is an essential element of resistance against aspergillus fumigatus. Nat Commun. 2021;12:3739.
Lu J, Marjon KD, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc receptors. Immunol Rev. 2012;250:230–8.
Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature. 2008;456:989–92.
Vilahur G, Badimon L. Biological actions of pentraxins. Vascul Pharmacol. 2015;73:38–44.
Clark SE, Weiser JN. Microbial modulation of host immunity with the small molecule phosphorylcholine. Infect Immun. 2013;81:392–401.
Medzhitov R, Janeway CA. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9.
Ullah MO, Sweet MJ, Mansell A, Kellie S, Kobe B. TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol. 2016;100:27–45.
Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90.
Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood. 2003;102:2660–9.
Parker LC, Whyte MKB, Dower SK, Sabroe I. The expression and roles of toll-like receptors in the biology of the human neutrophil. J Leukoc Biol. 2005;77:886–92.
Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD, DeLeo FR. Dectin-1 promotes fungicidal activity of human neutrophils. Eur J Immunol. 2007;37:467–78.
Li X, Utomo A, Cullere X, Choi MM, Milner DA, Venkatesh D, et al. The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe. 2011;10:603–15.
Kanneganti T-D, Lamkanfi M, Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–59.
Ekman A-K, Cardell LO. The expression and function of Nod-like receptors in neutrophils. Immunology. 2010;130:55–63.
Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16:35–50.
Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. 2012;12:215–25.
Kieser KJ, Kagan JC. Multi-receptor detection of individual bacterial products by the innate immune system. Nat Rev Immunol. 2017;17:376–90.
Peters-Sengers H, Butler JM, Uhel F, Schultz MJ, Bonten MJ, Cremer OL, et al. Source-specific host response and outcomes in critically ill patients with sepsis: a prospective cohort study. Intensive Care Med. 2022;48:92–102.
Liston A, Masters SL. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol. 2017;17:208–14.
Fu J, Wu H. Structural Mechanisms of NLRP3 Inflammasome assembly and activation. Annu Rev Immunol. 2023;41:301–16.
Linkermann A, Stockwell BR, Krautwald S, Anders H-J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–67.
Mantovani A, Garlanda C. Humoral innate immunity and acute-phase proteins. N Engl J Med. 2023;388:439–52.
Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82.
Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657–70.
Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102.
Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197–223.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5.
Abilés J, de la Cruz AP, Castaño J, Rodríguez-Elvira M, Aguayo E, Moreno-Torres R, et al. Oxidative stress is increased in critically ill patients according to antioxidant vitamins intake, independent of severity: a cohort study. Crit Care. 2006;10:R146.
Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–401.
Taneja R, Sharma AP, Hallett MB, Findlay GP, Morris MR. Immature circulating neutrophils in sepsis have impaired phagocytosis and calcium signaling. Shock. 2008;30:618–22.
Danikas DD, Karakantza M, Theodorou GL, Sakellaropoulos GC, Gogos CA. Prognostic value of phagocytic activity of neutrophils and monocytes in sepsis. correlation to CD64 and CD14 antigen expression. Clin Exp Immunol. 2008;154:87–97.
Mare TA, Treacher DF, Shankar-Hari M, Beale R, Lewis SM, Chambers DJ, et al. The diagnostic and prognostic significance of monitoring blood levels of immature neutrophils in patients with systemic inflammation. Crit Care. 2015;19:57.
Pöschl JMB, Ruef P, Linderkamp O. Deformability of passive and activated neutrophils in children with gram-negative septicemia. Scand J Clin Lab Invest. 2005;65:333–9.
Winkler MS, Rissiek A, Priefler M, Schwedhelm E, Robbe L, Bauer A, et al. Human leucocyte antigen (HLA-DR) gene expression is reduced in sepsis and correlates with impaired TNFα response: a diagnostic tool for immunosuppression? PLoS ONE. 2017;12: e0182427.
Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–5.
Calder PC. Eicosanoids. Essays Biochem. 2020;64:423–41.
Malavige GN, Ogg GS. Pathogenesis of vascular leak in dengue virus infection. Immunology. 2017;151:261–9.
Joffre J, Wong E, Lawton S, Lloyd E, Nguyen N, Xu F, et al. N-Oleoyl dopamine induces IL-10 via central nervous system TRPV1 and improves endotoxemia and sepsis outcomes. J Neuroinflammation. 2022;19:118.
Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101.
Lu C, Sharma S, McIntyre L, Rhodes A, Evans L, Almenawer S, et al. Omega-3 supplementation in patients with sepsis: a systematic review and meta-analysis of randomized trials. Ann Intensive Care. 2017;7:58.
Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. the Ibuprofen in sepsis study group. N Engl J Med. 1997;336:912–8.
Harkewicz R, Dennis EA. Applications of mass spectrometry to lipids and membranes. Annu Rev Biochem. 2011;80:301–25.
Watrous JD, Niiranen TJ, Lagerborg KA, Henglin M, Xu Y-J, Rong J, et al. Directed non-targeted mass spectrometry and chemical networking for discovery of eicosanoids and related oxylipins. Cell Chem Biol. 2019;26:433-442.e4.
Ogawa S, Gerlach H, Esposito C, Pasagian-Macaulay A, Brett J, Stern D. Hypoxia modulates the barrier and coagulant function of cultured bovine endothelium. increased monolayer permeability and induction of procoagulant properties. J Clin Invest. 1990;85:1090–8.
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83.
Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187:160–9.
Chiara AD, Pederzoli-Ribeil M, Burgel P-R, Danel C, Witko-Sarsat V. Targeting cytosolic proliferating cell nuclear antigen in neutrophil-dominated inflammation. Front Immunol. 2012;3:311.
Grégoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. 2018;52:1702590.
Bae H-B, Zmijewski JW, Deshane JS, Tadie J-M, Chaplin DD, Takashima S, et al. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J. 2011;25:4358–68.
Park DW, Jiang S, Liu Y, Siegal GP, Inoki K, Abraham E, et al. GSK3β-dependent inhibition of AMPK potentiates activation of neutrophils and macrophages and enhances severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L735-745.
van Gisbergen KPJM, Sanchez-Hernandez M, Geijtenbeek TBH, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med. 2005;201:1281–92.
Schuster S, Hurrell B, Tacchini-Cottier F. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J Leukoc Biol. 2013;94:671–5.
Gardner A, de Mingo PÁ, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol. 2020;11:924.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–66.
Seo G-Y, Giles DA, Kronenberg M. The role of innate lymphoid cells in response to microbes at mucosal surfaces. Mucosal Immunol. 2020;13:399–412.
Castellanos JG, Longman RS. The balance of power: innate lymphoid cells in tissue inflammation and repair. J Clin Investig. 2019;129:2640–50.
Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol. 2019;19:599–613.
Barlow JL, McKenzie ANJ. Innate lymphoid cells of the lung. Annu Rev Physiol. 2019;81:429–52.
Elemam NM, Ramakrishnan RK, Hundt JE, Halwani R, Maghazachi AA, Hamid Q. Innate lymphoid cells and natural killer cells in bacterial infections: function, dysregulation, and therapeutic targets. Front Cell Infect Microbiol. 2021;11: 733564.
Ebbo M, Crinier A, Vély F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol. 2017;17:665–78.
Mjösberg J, Spits H. Human innate lymphoid cells. J Allergy Clin Immunol. 2016;138:1265–76.
Silverstein NJ, Wang Y, Manickas-Hill Z, Carbone C, Dauphin A, Boribong BP, et al. Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. Elife. 2022;11:e74681.
Wang F, Cui Y, He D, Gong L, Liang H. Natural killer cells in sepsis: friends or foes? Front Immunol. 2023;14:1101918.
Pellicci DG, Koay H-F, Berzins SP. Thymic development of unconventional T cells: how NKT cells, MAIT cells and γδ T cells emerge. Nat Rev Immunol. 2020;20:756–70.
LeBlanc G, Kreissl FK, Melamed J, Sobel AL, Constantinides MG. The role of unconventional T cells in maintaining tissue homeostasis. Semin Immunol. 2022;61–64: 101656.
Jouan Y, Guillon A, Gonzalez L, Perez Y, Boisseau C, Ehrmann S, et al. Phenotypical and functional alteration of unconventional T cells in severe COVID-19 patients. J Exp Med. 2020;217: e20200872.
Flament H, Rouland M, Beaudoin L, Toubal A, Bertrand L, Lebourgeois S, et al. Outcome of SARS-CoV-2 infection is linked to MAIT cell activation and cytotoxicity. Nat Immunol. 2021;22:322–35.
Parrot T, Gorin J-B, Ponzetta A, Maleki KT, Kammann T, Emgård J, et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci Immunol. 2020. https://doi.org/10.1126/sciimmunol.abe1670.
Legoux F, Salou M, Lantz O. Unconventional or preset αβ T cells: evolutionarily conserved tissue-resident T cells recognizing nonpeptidic ligands. Annu Rev Cell Dev Biol. 2017;33:511–35.
Davey MS, Willcox CR, Hunter S, Oo YH, Willcox BE. Vδ2+ T cells—two subsets for the price of one. Front Immunol. 2018;9:2106.
Godfrey DI, Koay H-F, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol. 2019;20:1110–28.
Crosby CM, Kronenberg M. Tissue-specific functions of invariant natural killer T cells. Nat Rev Immunol. 2018;18:559–74.
Grimaldi D, Le Bourhis L, Sauneuf B, Dechartres A, Rousseau C, Ouaaz F, et al. Specific MAIT cell behaviour among innate-like T lymphocytes in critically ill patients with severe infections. Intensive Care Med. 2014;40:192–201.
van Wilgenburg B, Scherwitzl I, Hutchinson EC, Leng T, Kurioka A, Kulicke C, et al. MAIT cells are activated during human viral infections. Nat Commun. 2016;7:11653.
Hackstein C-P, Klenerman P. Emerging features of MAIT cells and other unconventional T cell populations in human viral disease and vaccination. Semin Immunol. 2022;61–64: 101661.
Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T cell targets for cancer immunotherapy. Immunity. 2018;48:453–73.
Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45.
Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. 2021;32:314–24.
Chavakis T, Santoso S, Clemetson KJ, Sachs UJH, Isordia-Salas I, Pixley RA, et al. High molecular weight kininogen regulates platelet-leukocyte interactions by bridging Mac-1 and glycoprotein Ib. J Biol Chem. 2003;278:45375–81.
Meziani F, Tesse A, Andriantsitohaina R. Microparticles are vectors of paradoxical information in vascular cells including the endothelium: role in health and diseases. Pharmacol Rep. 2008;60:75–84.
Delabranche X, Berger A, Boisramé-Helms J, Meziani F. Microparticles and infectious diseases. Med Mal Infect. 2012;42:335–43.
Joffre J, Hellman J. Oxidative stress and endothelial dysfunction in sepsis and acute inflammation. Antioxid Redox Signal. 2021;35:1291–307.
Stiel L, Meziani F, Helms J. Neutrophil activation during septic shock. Shock. 2018;49:371–84.
Kvietys PR, Fakhoury HMA, Kadan S, Yaqinuddin A, Al-Mutairy E, Al-Kattan K. COVID-19: lung-centric immunothrombosis. Front Cell Infect Microbiol. 2021;11: 679878.
Lim MS, Mcrae S. COVID-19 and immunothrombosis: pathophysiology and therapeutic implications. Crit Rev Oncol Hematol. 2021;168: 103529.
Portier I, Campbell RA, Denorme F. Mechanisms of immunothrombosis in COVID-19. Curr Opin Hematol. 2021;28:445–53.
Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: cross talks between inflammation and coagulation. Respiration. 2017;93:212–25.
Zhang H, Goswami J, Varley P, van der Windt DJ, Ren J, Loughran P, et al. Hepatic surgical stress promotes systemic immunothrombosis that results in distant organ injury. Front Immunol. 2020;11:987.
Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S, et al. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget. 2018;9:1772–84.
Netea MG, van der Meer JWM. Trained immunity: an ancient way of remembering. Cell Host Microbe. 2017;21:297–300.
Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086.
Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Jacobs C, Xavier RJ, et al. BCG-induced trained immunity in NK cells: role for non-specific protection to infection. Clin Immunol. 2014;155:213–9.
Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–32.
Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A. 2012;109:17537–42.
Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T, et al. Trained immunity, tolerance, priming and differentiation: distinct immunological processes. Nat Immunol. 2021;22:2–6.
Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 2018;172:176-190.e19.
Khan N, Downey J, Sanz J, Kaufmann E, Blankenhaus B, Pacis A, et al. tuberculosis reprograms hematopoietic stem cells to limit myelopoiesis and impair trained immunity. Cell. 2020. https://doi.org/10.1016/j.cell.2020.09.062.
van der Heijden WA, Van de Wijer L, Keramati F, Trypsteen W, Rutsaert S, Horst RT, et al. Chronic HIV infection induces transcriptional and functional reprogramming of innate immune cells. JCI Insight. 2021;6: 145928.
Hong M, Sandalova E, Low D, Gehring AJ, Fieni S, Amadei B, et al. Trained immunity in newborn infants of HBV-infected mothers. Nat Commun. 2015;6:6588.
Schrum JE, Crabtree JN, Dobbs KR, Kiritsy MC, Reed GW, Gazzinelli RT, et al. Cutting edge: plasmodium falciparum induces trained innate immunity. J Immunol. 2018;200:1243–8.
Stevens NE, van Wolfswinkel M, Bao W, Ryan FJ, Brook B, Amenyogbe N, et al. Immunisation with the BCG and DTPw vaccines induces different programs of trained immunity in mice. Vaccine. 2022;40:1594–605.
Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172:162-175.e14.
Zhang H, Chen T, Ren J, Xia Y, Onuma A, Wang Y, et al. Pre-operative exercise therapy triggers anti-inflammatory trained immunity of Kupffer cells through metabolic reprogramming. Nat Metab. 2021;3:843–58.
de Bree LCJ, Mourits VP, Koeken VA, Moorlag SJ, Janssen R, Folkman L, et al. Circadian rhythm influences induction of trained immunity by BCG vaccination. J Clin Invest. 2020;130:5603–17.
Arts RJW, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–19.
Domínguez-Andrés J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, Arts RJW, et al. The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab. 2019;29:211-220.e5.
Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. 2018;172:135-146.e9.
Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018;172:147-161.e12.
Rivas MN, Ebinger JE, Wu M, Sun N, Braun J, Sobhani K, et al. BCG vaccination history associates with decreased SARS-CoV-2 seroprevalence across a diverse cohort of health care workers. J Clin Invest. 2021;131: 145157.
Chumakov K, Avidan MS, Benn CS, Bertozzi SM, Blatt L, Chang AY, et al. Old vaccines for new infections: exploiting innate immunity to control COVID-19 and prevent future pandemics. Proc Natl Acad Sci U S A. 2021;118: e2101718118.
Ospelt C, Reedquist KA, Gay S, Tak PP. Inflammatory memories: is epigenetics the missing link to persistent stromal cell activation in rheumatoid arthritis? Autoimmun Rev. 2011;10:519–24.
Herz U, Gerhold K, Grüber C, Braun A, Wahn U, Renz H, et al. BCG infection suppresses allergic sensitization and development of increased airway reactivity in an animal model. J Allergy Clin Immunol. 1998;102:867–74.
Ochando J, Fayad ZA, Madsen JC, Netea MG, Mulder WJM. Trained immunity in organ transplantation. Am J Transplant. 2020;20:10–8.
Casanova J-L, Abel L. Mechanisms of viral inflammation and disease in humans. Science. 2021;374:1080–6.
Hodeib S, Herberg JA, Levin M, Sancho-Shimizu V. Human genetics of meningococcal infections. Hum Genet. 2020;139:961–80.
O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature. 2021;590:140–5.
Zhang Q, Matuozzo D, Le Pen J, Lee D, Moens L, Asano T, et al. Recessive inborn errors of type I IFN immunity in children with COVID-19 pneumonia. J Exp Med. 2022;219: e20220131.
Hoste L, Van Paemel R, Haerynck F. Multisystem inflammatory syndrome in children related to COVID-19: a systematic review. Eur J Pediatr. 2021;180:2019–34.
Lee D, Le Pen J, Yatim A, Dong B, Aquino Y, Ogishi M, et al. Inborn errors of OAS-RNase L in SARS-CoV-2-related multisystem inflammatory syndrome in children. Science. 2023. https://doi.org/10.1126/science.abo3627.
Bline KE, Hall MW. Immune function in critically Ill septic children. Pathogens. 2021;10:1239.
Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med. 2011;37:525–32.
Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, et al. Stereotypic immune system development in newborn children. Cell. 2018;174:1277-1292.e14.
Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity. 2017;46:350–63.
Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–89.
Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583–93.
Sauce D, Dong Y, Campillo-Gimenez L, Casulli S, Bayard C, Autran B, et al. Reduced oxidative burst by primed neutrophils in the elderly individuals is associated with increased levels of the cd16bright/cd62ldim immunosuppressive subset. J Gerontol A Biol Sci Med Sci. 2017;72:163–72.
Fulop T, Larbi A, Douziech N, Fortin C, Guérard K-P, Lesur O, et al. Signal transduction and functional changes in neutrophils with aging. Aging Cell. 2004;3:217–26.
Nyugen J, Agrawal S, Gollapudi S, Gupta S. Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol. 2010;30:806–13.
Hearps AC, Martin GE, Angelovich TA, Cheng W-J, Maisa A, Landay AL, et al. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell. 2012;11:867–75.
De Martinis M, Modesti M, Ginaldi L. Phenotypic and functional changes of circulating monocytes and polymorphonuclear leucocytes from elderly persons. Immunol Cell Biol. 2004;82:415–20.
van Duin D, Mohanty S, Thomas V, Ginter S, Montgomery RR, Fikrig E, et al. Age-associated defect in human TLR-1/2 function. J Immunol. 2007;178:970–5.
Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell. 2014;13:699–708.
Hilmer SN, Cogger VC, Le Couteur DG. Basal activity of Kupffer cells increases with old age. J Gerontol A Biol Sci Med Sci. 2007;62:973–8.
Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res. 2012;32:18–26.
Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. 2017;21:455-466.e4.
Panda A, Qian F, Mohanty S, van Duin D, Newman FK, Zhang L, et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol. 2010;184:2518–27.
D’Souza SS, Shen X, Fung ITH, Ye L, Kuentzel M, Chittur SV, et al. Compartmentalized effects of aging on group 2 innate lymphoid cell development and function. Aging Cell. 2019. https://doi.org/10.1111/acel.13019.
Lee O-J, Cho Y-N, Kee S-J, Kim M-J, Jin H-M, Lee S-J, et al. Circulating mucosal-associated invariant T cell levels and their cytokine levels in healthy adults. Exp Gerontol. 2014;49:47–54.
Colonna-Romano G, Aquino A, Bulati M, Lio D, Candore G, Oddo G, et al. Impairment of gamma/delta T lymphocytes in elderly: implications for immunosenescence. Exp Gerontol. 2004;39:1439–46.
Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–22.
Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and “Garb-aging.” Trends Endocrinol Metab. 2017;28:199–212.
Dall’Olio F, Vanhooren V, Chen CC, Slagboom PE, Wuhrer M, Franceschi C. N-glycomic biomarkers of biological aging and longevity: a link with inflammaging. Ageing Res Rev. 2013;12:685–98.
Biagi E, Candela M, Fairweather-Tait S, Franceschi C, Brigidi P. Aging of the human metaorganism: the microbial counterpart. Age (Dordr). 2012;34:247–67.
Walker KA, Basisty N, Wilson DM, Ferrucci L. Connecting aging biology and inflammation in the omics era. J Clin Invest. 2022;132: e158448.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7.
Conte M, Martucci M, Chiariello A, Franceschi C, Salvioli S. Mitochondria, immunosenescence and inflammaging: a role for mitokines? Semin Immunopathol. 2020;42:607–17.
Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88.
Martínez G, Duran-Aniotz C, Cabral-Miranda F, Vivar JP, Hetz C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell. 2017;16:615–23.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4-9.
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol. 2017;8:1960.
Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med. 2006;34:15–21.
Martin-Loeches I, Guia MC, Vallecoccia MS, Suarez D, Ibarz M, Irazabal M, et al. Risk factors for mortality in elderly and very elderly critically ill patients with sepsis: a prospective, observational, multicenter cohort study. Ann Intensive Care. 2019;9:26.
Lu X, Yang Y-M, Lu Y-Q. Immunosenescence: a critical factor associated with organ injury after sepsis. Front Immunol. 2022;13: 917293.
Monneret G, Gossez M, Venet F. Sepsis and immunosenescence: closely associated in a vicious circle. Aging Clin Exp Res. 2021;33:729–32.
Liu D, Huang S-Y, Sun J-H, Zhang H-C, Cai Q-L, Gao C, et al. Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Mil Med Res. 2022;9:56.
Verhoef G, Boogaerts M. Treatment with granulocyte-macrophage colony stimulating factor and the adult respiratory distress syndrome. Am J Hematol. 1991;36:285–7.
Roquilly A, Francois B, Huet O, Launey Y, Lasocki S, Weiss E, et al. Interferon gamma-1b for the prevention of hospital-acquired pneumonia in critically ill patients: a phase 2, placebo-controlled randomized clinical trial. Intensive Care Med. 2023;49:530–44.
Burnham KL, Davenport EE, Radhakrishnan J, Humburg P, Gordon AC, Hutton P, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. 2017;196:328–39.
Sweeney TE, Azad TD, Donato M, Haynes WA, Perumal TM, Henao R, et al. Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit Care Med. 2018;46:915–25.
Wong HR, Sweeney TE, Hart KW, Khatri P, Lindsell CJ. Pediatric sepsis endotypes among adults with sepsis. Crit Care Med. 2017;45:e1289–91.
Antcliffe DB, Burnham KL, Al-Beidh F, Santhakumaran S, Brett SJ, Hinds CJ, et al. Transcriptomic signatures in sepsis and a differential response to steroids. from the VANISH randomized trial. Am J Respir Crit Care Med. 2019;199:980–6.
Bendib I, Beldi-Ferchiou A, Schlemmer F, Surenaud M, Maitre B, Plonquet A, et al. Alveolar compartmentalization of inflammatory and immune cell biomarkers in pneumonia-related ARDS. Crit Care. 2021;25:23.
Jouan Y, Baranek T, Si-Tahar M, Paget C, Guillon A. Lung compartmentalization of inflammatory biomarkers in COVID-19-related ARDS. Crit Care. 2021;25:120.
Conway Morris A, Rynne J, Shankar-Hari M. Compartmentalisation of immune responses in critical illness: does it matter? Intensive Care Med. 2022;48:1617–20.
Doherty GM, Lange JR, Langstein HN, Alexander HR, Buresh CM, Norton JA. Evidence for IFN-gamma as a mediator of the lethality of endotoxin and tumor necrosis factor-alpha. J Immunol. 1992;149:1666–70.
Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184:149-168.e17.
Kotsaki A, Pickkers P, Bauer M, Calandra T, Lupse M, Wiersinga WJ, et al. ImmunoSep (Personalised Immunotherapy in Sepsis) international double-blind, double-dummy, placebo-controlled randomised clinical trial: study protocol. BMJ Open. 2022;12: e067251.
Leventogiannis K, Kyriazopoulou E, Antonakos N, Kotsaki A, Tsangaris I, Markopoulou D, et al. Toward personalized immunotherapy in sepsis: the PROVIDE randomized clinical trial. Cell Rep Med. 2022;3: 100817.
Acknowledgement
Figures were created with BioRender.com
Funding
Not applicable.
Author information
Authors and Affiliations
Consortia
Contributions
All the authors met authorship criteria and participated significantly to the study; in particular, LS, AG, and YR were involved in conception and design; LS, AG, YJ, HV, ST, PB wrote the article; JJ, GV, NB, BS, SB, MO, HKand LK provided critical revisions to the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stiel, L., Gaudet, A., Thietart, S. et al. Innate immune response in acute critical illness: a narrative review. Ann. Intensive Care 14, 137 (2024). https://doi.org/10.1186/s13613-024-01355-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13613-024-01355-6