
Abstract
Background
Primary brain rhabdomyosarcoma is a rare primary brain malignancy with few case reports. The vast majority of cases of primary brain rhabdomyosarcoma occur in pediatric patients, and immunohistochemistry can distinguish it from embryonal subtypes; however, few cases of primary brain rhabdomyosarcoma in adults have been reported in the literature.
Case presentation
We report the case of a 26-year-old White male patient who was found to have primary brain alveolar rhabdomyosarcoma after developing headaches for several months. A brain MRI revealed a mixed cystic and solid tumor along the vermis of the cerebellum. The patient underwent a gross total surgical resection, which confirmed the diagnosis of alveolar rhabdomyosarcoma. Further staging workup for another primary focus or disseminated disease yielded negative results, confirming the diagnosis of primary alveolar rhabdomyosarcoma of the brain.
Conclusion
The standard of care for managing this rare type of brain tumor involves surgery with adjuvant chemoradiotherapy. Further studies should be conducted for a better diagnostic and therapeutic understanding.
Similar content being viewed by others


Introduction
Primary brain rhabdomyosarcoma is a rare primary brain malignancy with scant case reports. While the majority of cases of primary brain rhabdomyosarcoma occur in pediatric patients, a few cases of primary brain rhabdomyosarcoma in adults have been reported in the literature. Myogenin immunostaining has been described as a useful marker of the alveolar subtype of rhabdomyosarcoma and as a tool for distinguishing it from the more common embryonal subtype [1]. We report a rare case of an adult who was diagnosed with primary brain alveolar rhabdomyosarcoma, for which an extensive immunohistochemistry workup and molecular profiling panel were done.
Case report
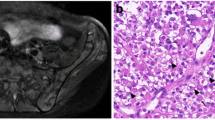
The patient was a 26-year-old White male with no known medical history who started to complain of headaches six months before presentation. The headache worsened over time and became more severe in the last month. No abnormal findings on physical examination were noted. No nausea, vomiting, unsteadiness, or any other associated signs or symptoms. An MRI of the brain (Fig. 1) showed a 4.5 × 4.3 × 3.9 cm mixed solid and cystic tumor along the surface of the vermis of the cerebellum. It contained multiple cystic areas with blood and a solid, strongly enhancing component. There was no surrounding vasogenic edema. It caused a mass effect on the fourth ventricle with secondary supratentorial hydrocephalus and CSF seepage. There was cerebellar tonsillar herniation and upward herniation of the cerebellum, with effacement of the quadrigeminal cistern. The radiological findings were suggestive of an ependymoma. An MRI of the spine was performed to complete the workup and revealed no abnormalities.

a axial non-enhanced T1, b axial enhanced T1 weighted image and c. axial FLAIR image of the brain. There is a predominantly T1 hypointense mass along the vermis, containing areas of T1 hyperintense fluid–fluid level. It shows strong heterogenous enhancement after contrast administration (b). There is no significant surrounding edema on FLAIR (c). It is compressing and displacing the fourth ventricle anteriorly
After case discussion and imaging review in a multidisciplinary meeting at our institution involving oncology, neurosurgery, pathology, radiation oncology, and radiology, the patient underwent surgery and gross total resection of the tumor after one week of presentation. Surprisingly, the pathology showed a high-grade neoplasm with immunophenotypic and molecular features of alveolar rhabdomyosarcoma. Histologic examination revealed a densely cellular neoplasm comprising round cells arranged in sheets. The tumor cells had round-to-oval nuclei and prominent nucleoli (Fig. 2A, B). The mitotic figures were elevated, and scattered foci of necrosis were identified. Entrapped islands of the neuropil were noted focally. Immunohistochemistry revealed that the tumor cells were negative for the glial markers GFAP and OLIG2. Desmin and myogenin were strongly expressed in a subset of tumor cells (Fig. 2C, D). The cells showed variable expression levels of BCOR, BCL2, BCL6, p53, INSM1, and synaptophysin. They were negative for BRAF V600E mutant protein, CAM5.2, EMA, H3K27M mutant protein, mutant IDH1, NeuN, S100, and SOX-10. H3K27 trimethylation was preserved. In addition, the nuclear expression of INI-1 and ATRX was retained. The Ki-67 proliferative index was elevated. A molecular oncogenic gene fusion panel detected a PAX3-INO80D fusion. Two PTEN mutations (p.V290fs and p.T319*) were identified using Next Generation sequencing (NGS). There were no mutations in IDH1, IDH2, BRAF, TP53, histone H3, NF1, NF2, DICER1, RB1, CDKN2A, ATRX, or TERT promoters. There were no reportable copy numbers. The overall findings were those of a high-grade neoplasm with immunophenotypic and molecular findings consistent with alveolar rhabdomyosarcoma. The staging workup, which included total-body CT scans and a testicular ultrasound, was negative for the primary disease. Therefore, we are dealing with a rare entity among adults called primary alveolar rhabdomyosarcoma of the brain. Our patient was scheduled to start radiation therapy in his home country, followed by systemic therapy (VDC-IE protocol). The patient was then lost to follow-up.

Alveolar rhabdomyosarcoma. A and B (H&E stain). The tumor is hypercellular and is composed of round cells with prominent nucleoli. Mitotic activity is brisk. C The tumor cells show strong positivity for Desmin. D Myogenin is positive in a subset of tumor cells
Discussion
Rhabdomyosarcomas are common tumors of the head and neck region in children and rarely in adults. In addition, the intracranial localization of this tumor is rare. Among the few primary brain sarcomas, 70% arise in the pediatric population, and there have been limited cases reported in adults [2]. Primary intracranial sarcomas account for only 0.1–4.3% of all intracranial neoplasms and are more likely to be found in children [3]. Further classification of brain rhabdomyosarcoma can be based on the cell of origin, where intracranial rhabdomyosarcomas are more likely to arise from the brain substance and extend to the meninges, in contrast to meningeal rhabdomyosarcomas that arise from pluripotent mesenchymal cells of the meninges [4].
The majority of case reports in the literature describe cases of brain rhabdomyosarcoma of secondary origin, such as from the extremities, lungs, scalp, and genital tract. A few cases of primary intracranial alveolar rhabdomyosarcoma have been reported. A review of the literature identified 47 cases of primary cerebral rhabdomyosarcoma, of which 33 were children, and the majority were embedded in the surface of the brain and rarely entirely intraparenchymal or intraventricular [5]. Our patient was a rare case of primary cerebellar rhabdomyosarcoma in an adult patient.
The clinical and histological criteria currently used to classify rhabdomyosarcomas are complex, and their predictive power is limited. However, the role of molecular assays in the subclassification of patients with alveolar rhabdomyosarcoma by associating distinctive clinical patterns with the common and variant gene fusions has been shown to be helpful. For example, alveolar rhabdomyosarcoma is characterized by consistent chromosomal translocations and chimeric genes, PAX3-FKHR and PAX7-FKHR, which are expressed as novel fusion transcripts [6].
Following histological diagnosis and staging, there is still much debate regarding the optimal course of treatment. The inherent difficulty in discerning the cell of origin in rhabdomyosarcoma makes the selection of appropriate chemotherapy difficult and its usefulness debatable [7]. In addition, given the rarity of intracranial rhabdomyosarcoma and the heterogeneity of treatments, little could be concluded regarding the best therapeutic approach. However, the most common approach is to perform surgery first, followed by adjuvant radiation therapy and chemotherapy.
Conclusion
Our patient is one of the few adults with primary brain rhabdomyosarcoma. Given the rarity of these tumors, a multidisciplinary approach is necessary. Currently, the safest surgical resection of the tumor, followed by adjuvant chemotherapy and radiotherapy, remains the logical approach to managing these tumors to maximize long-term survival. However, further studies are required to better understand the behavior of these tumors, identify an optimal therapeutic plan and standardize diagnostic immunohistochemistry and molecular profiling tests.
Availability of data and materials
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to patient confidentiality pertaining to medical electronic health records from which our data were collected.
References
Morgenstern D, Rees H, Sebire N, Shipley J, Anderson J. Rhabdomyosarcoma subtyping by immunohistochemical assessment of myogenin: tissue array study and review of the literature. Pathol Oncol Res. 2008;14:233–8.
Dropcho EJ, Allen JC. Primary intracranial rhabdomyosarcoma: case report and review of the literature. J Neurooncol. 1987;5(2):139–50.
Al-Gahtany M, Shroff M, Bouffet E, Dirks P, Drake J, Humphreys R, et al. Primary central nervous system sarcomas in children: clinical, radiological, and pathological features. Childs Nerv Syst. 2003;19(12):808–17.
Xu F, De Las Casas LE, Dobbs LJ Jr. Primary meningeal rhabdomyosarcoma in a child with hypomelanosis of Ito. Arch Pathol Lab Med. 2000;124(5):762–5.
Khalatbari MR, Hamidi M, Moharamzad Y. Primary alveolar rhabdomyosarcoma of the brain with long-term survival. J Neurooncol. 2013;115(1):131–3.
Kelly KM, Womer RB, Sorensen PH, Xiong QB, Barr FG. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol. 1997;15(5):1831–6.
Palta M, Riedel RF, Vredenburgh JJ, Cummings TJ, Green S, Chang Z, et al. Primary meningeal rhabdomyosarcoma. Sarcoma. 2011;2011: 312802.
Acknowledgements
Not applicable.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
MN and HA conceived the idea for the paper. LAM and NC drafted the original manuscript. RH, AT and SS contributed to the discussion section. RAZ and GA reviewed and edited the final manuscript. HA and BY supervised the project. All authors contributed, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors complied with the ethical requirements of their institution.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Al Mahmasani, L., Najjar, M., Hourany, R. et al. Primary alveolar rhabdomyosarcoma of the brain: a case report. J Med Case Reports 18, 178 (2024). https://doi.org/10.1186/s13256-024-04471-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-024-04471-w