
Abstract
Background and purpose
Early detection, diagnosis, and treatment of colorectal cancer and its precancerous lesions can significantly improve patients’ survival rates. The purpose of this research is to identify methylation markers specific to colorectal cancer tissues and validate their diagnostic capability in colorectal cancer and precancerous changes by measuring the level of DNA methylation in stool samples.
Method
We analyzed samples from six cancer tissues and adjacent normal tissues and fecal samples from 758 participants, including 62 patients with interfering diseases. Bioinformatics databases were used to screen for candidate biomarkers for CRC, and quantitative methylation-specific PCR methods were applied for identification. The methylation levels of the candidate biomarkers in fecal and tissue samples were measured. Logistic regression and random forest models were built and validated using fecal sample data from one of the centers, and the independent or combined diagnostic value of the candidate biomarkers in fecal samples for CRC and precancerous lesions was analyzed. Finally, the diagnostic capability and stability of the model were validated at another medical center.
Results
This study identified two colorectal cancer CpG sites with tissue specificity. These two biomarkers have certain diagnostic power when used individually, but their diagnostic value for colorectal cancer and colorectal adenoma is more significant when they are used in combination.
Conclusion
The results indicate that a DNA methylation biomarker combined diagnostic model based on two CpG sites, cg13096260 and cg12587766, has the potential for screening and diagnosing precancerous lesions and colorectal cancer. Additionally, compared to traditional diagnostic models, machine learning algorithms perform better but may yield more false-positive results, necessitating further investigation.
Similar content being viewed by others


Background
Colorectal cancer is a highly prevalent malignant tumor worldwide, posing a serious threat to human health and has become one of the major global public health issues [1]. Like other tumors, early detection, diagnosis, and treatment of the disease can significantly improve the prognosis of patients [1, 2]. Therefore, early detection will help improve the survival rate and quality of life of patients with colorectal cancer. However, clinical success in developing effective, noninvasive or minimally invasive diagnostic methods is still relatively limited.
The incidence of colorectal cancer is showing a trend toward younger ages, and the number of late-stage cases at initial diagnosis is gradually increasing, forcing us to raise higher demands for early screening and diagnostic methods [3]. The development of colorectal cancer mainly involves cumulative genetic and epigenetic changes, and it usually takes 10 to 15 years for precancerous lesions to develop into CRC [4, 5]. This provides a theoretical basis and the best window for screening by identifying tumor-specific changes through the analysis of DNA from exfoliated cells in feces. Numerous studies have confirmed that effective screening methods can help detect and remove precancerous lesions and early colorectal cancer, thereby reducing the incidence and mortality of colorectal cancer. Fecal occult blood tests and endoscopic examinations of the digestive system are currently commonly used as clinical screening tools for CRC [6, 7]. However, due to the low participation rate of traditional screening methods and the limitations of screening tool performance, large-scale screening work has become difficult [8]. Therefore, it is necessary to adopt more effective screening strategies and diagnostic methods to strengthen secondary prevention measures for colorectal cancer.
The progression of colorectal cancer is intimately associated with epigenetic mechanisms, with aberrant DNA methylation as one of its central characteristics [9,10,11,12]. It is typically tightly linked to gene expression defects, resulting in an imbalance between the expression of proto-oncogenes and tumor suppressor genes. The increase in methylation of tumor-related genes and the decrease in the degree of whole-genome methylation are early events in various types of colorectal cancer. Recent research has found that abnormal methylation may have occurred before the tumor progresses to the adenoma stage, which may be one of the earliest detectable tumor changes [13,14,15]. David et al. diagnosed adenomas and CRC by detecting abnormal methylation changes in the vimentin, NDRG4, BMP3, and TFPI2 genes in the DNA of exfoliated cells in feces. The research results showed that for patients with colorectal cancer, the sensitivity of this method is 85%, for adenomas (≥ 1 cm), the sensitivity is 54%, and the specificity is 90% [16]. Concurrently, Imperiale and colleagues incorporated 9989 asymptomatic individuals in a similar study involving multi-target stool DNA (sDNA) methylation and fecal immunochemical testing. The results demonstrated that the sensitivity of this method for colorectal cancer was 92.3%, the sensitivity for advanced precancerous changes was 42.4%, the specificity was 86.6%, and its screening performance was notably superior to the standard FIT [17]. Based on this research, the U.S. Food and Drug Administration approved the first sDNA methylation detection kit, Cologuard™ (Exact Science, Madison WI), for early screening of CRC. However, the diagnostic ability for early lesions has not yet met the demand, and the impact of racial differences is not yet clear. Therefore, further research is needed on the potential value of sDNA carrying cancer-specific methylation genes for CRC screening and early diagnosis.
SDC2 belongs to the syndecan family and is highly expressed in various cancers, including osteosarcoma, breast cancer, and colorectal cancer. High methylation of the SDC2 promoter region is a common epigenetic change in the development of colorectal tumors. Numerous methylation alterations in the SDC2 gene can be identified in the stool of patients with early-stage colorectal cancer and advanced adenomas, rendering it a potential new target for early detection. The leukemia inhibitory factor receptor (LIFR) is part of the type I cytokine receptor family, plays a role in promoting stem cell pluripotency, regulating cell proliferation and differentiation, and is also overexpressed in a range of tumor tissues [18,19,20]. Additionally, research has discovered a high methylation status in the promoter region of LIFR-AS1 in colorectal cancer, and according to in vitro and in vivo studies, its overexpression can significantly suppress the proliferation, growth, and invasive phenotype of colon cancer cells [21]. However, because SDC2 and LIFR are also highly expressed in other tumors, this may lead to false-positive diagnoses of other types of cancer. Since cancers from different tissue types may have similar methylation changes, methylation markers used for screening or cancer diagnosis in asymptomatic populations should have tissue specificity [22]. If lacking of tissue specificity, it is impossible to determine the source of the tumor and choose subsequent diagnostic methods.
Hence, our study is designed to pinpoint methylation markers that are specifically expressed in colorectal cancer via bioinformatics analysis. Then, analyze the clinical performance of these specifically expressed methylation markers in CRC detection, diagnosis of precancerous lesions, and other interfering diseases. Subsequently, we identified CRC methylation markers in sDNA with high specificity and sensitivity and separately established logistic diagnostic models and random forest diagnostic models. In the end, the stability and diagnostic effectiveness of these models were validated in an external cohort.
Methods
Study design
The purpose of this study is to explore and validate new biomarkers with tissue-specific methylation by analyzing biological databases, utilizing pyrosequencing of colorectal cancer tissues, and CpG sites in the DNA of exfoliated cells in feces. Then evaluate the impact of different modeling methods such as conventional modeling methods and machine learning methods on diagnostic performance. Then, we employ the constructed model for external validation in the dataset of a different medical center, and evaluate the stability of the model across various medical centers. This research has received approval from the Human Research Ethics Committee of the Sichuan Provincial People’s Hospital, and all participants have given their written informed consent (accession nos: Ethical Application Review (Research) 2022 No. 306).
Patient and sample collection
The DNA methylation data for screening tissue-specific methylation sites mainly come from TCGA (including TCGA-COAD and TCGA-READ) and published literatures. The DNA methylation data for verifying tissue-specific methylation sites come from the Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital and Shanghai Bohao Biotechnology Co., Ltd. Complete clinical, molecular, and histopathological datasets can be obtained from the Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, Chongqing Bohao Diagnostic Technology Co., Ltd., and the TCGA website (https://tcga-data.nci.nih.gov/tcga/). Both the dual-center data of this study and the TCGA dataset use the same platform (Illumina) for methylation status analysis.
This study collected samples from 6 cases of cancer tissues and adjacent normal tissues and 758 fecal samples from CRC patients, AA patients, patients with interfering diseases (including benign disease patients and other tumor disease patients), and healthy individuals. The tissue samples were cut into tissue blocks with a maximum thickness of no more than 0.5 cm on any side within 30 min after being removed from the body, and then immediately placed in 2 ml DNA preservation solution. The tissue preservation solution was left overnight in a 4 °C environment, and then transferred to a − 20 °C environment for long-term storage. Stool samples were collected from patients without bowel preparation, each sample weighing approximately 5 g, and then placed in a 50 ml centrifuge tube containing 25 ml preservation solution. Stool samples were stored at − 20 °C for a long time before use. These samples were collected from the Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital and Chongqing Bohao Diagnostic Technology Co., Ltd.
The inclusion criteria for this study are as follows: (1) voluntary participation in the study and signing of informed consent; (2) age ≥ 18 years, both sexes; (3) pathologically confirmed as colorectal cancer at different stages; (4) no preoperative radiation, chemotherapy or molecular-target or immuno therapy, and no history of other serious diseases. The exclusion criteria for this study are as follows: (1) pregnant or breastfeeding women; (2) women of childbearing age who test positive for pregnancy at baseline; (3) have had serious cardiovascular diseases within 12 months before enrollment, such as symptomatic coronary heart disease, congestive heart failure ≥ grade II, uncontrolled arrhythmia, myocardial infarction; (4) concurrent severe uncontrolled infections or other severe uncontrolled comorbidities, moderate or severe kidney injury; (5) not pregnant, breastfeeding women or women of childbearing age who test positive for pregnancy. The following clinical information of the participants were collected, including age, gender, tumor size, some tumor indicators, tumor location, histological type, lymphatic invasion, distant metastasis, pathological staging (determined according to the AJCC 8th edition TNM tumor staging system), vascular invasion, nerve invasion, microsatellite status, and some gene mutation situations.
Identification and selection criteria for methylation markers
To determine the candidate markers, we performed a bioinformatics analysis of the methylation data in the public databases (TCGA, TCGA-COAD, and TCGA-READ) and screened out the candidate tissue-specific sites. These methylation data mainly include Infinium Human Methylation 450 BeadChip DNA methylation data (Illumina, San Diego, CA, USA), which covers tumor tissues from 387 CRC patients and colon tissues from 45 healthy individuals. Subsequently, the DNA methylation levels of specific CpG sites in the candidate genes were validated by pyrosequencing of colorectal cancer tissues. Then, in two retrospective sDNA cohorts, the expression of the selected colorectal cancer methylation markers in fecal samples was detected using quantitative methylation-specific PCR (qMSP).
Verification of methylation status of tissue-paired specimens by pyrosequencing
The DNA methylation levels of specific CpG sites in candidate genes were quantitatively assessed by pyrosequencing. The median CpG value in each sample represents the DNA methylation status of each gene. Primers are designed using PyroMark Assay Design 2.0 software and synthesized by Bioengineering (Shanghai) Co., Ltd. Primer sequences and primer group information can be found in Supplementary File 1: Table S1. Pyrosequencing reactions and quantification of DNA methylation were performed on the Pyromark Q96 MD pyrosequencing system (QIAGEN, Valencia, CA, USA).
We have taken the following quality control measures to ensure the accuracy and authenticity of the pyrosequencing results. Firstly, the quality of the extracted DNA was detected and evaluated using 1% agarose gel electrophoresis. Then, a sample group of serial dilutions of fully methylated and non-methylated DNA (Human Methylated and Non-methylated DNA Set, Zymo Research, Freiburg im Breisgau, Germany) (0%, 25%, 50%, 75%, and 100%) was used as a negative and positive control in methylation detection applications against DNA standards. In addition, the standard and the sample were tested simultaneously to evaluate the conversion efficiency of bisulfite. Lastly, it was ensured that all samples were mixed in different culture plates, and no template control was included in each run of the experiment.
DNA capture and bisulfite conversion
First, the fecal samples are ground into a uniform slurry with glass beads, then centrifuged at 4000×g for 10 min, the supernatant is taken and this step is repeated. Finally, sDNA is extracted using the MagicPure® Fecal and Soil Genomic DNA Kit (Full-Form Gold EC801-11, Beijing, China). The extracted supernatant is stored at − 20 °C. The extracted DNA samples (at least 10 ng) are converted and purified with bisulfite, using the EZ DNA Methylation-Lightning Kit (ZYMO RESEARCH, D5031, Los Angeles, CA, USA). The concentration of the extracted DNA is quantified using a spectrophotometer, and then the samples are stored at − 20 °C for subsequent use.
Detection of quantitative methylation-specific PCR
Quantitative methylation-specific PCR (qMSP) is used to quantitatively detect the methylation status of SDC2 (including gl3096260), LIFR (including cg12587766), and ACTB genes in fecal samples, and ACTB is amplified as an internal reference for DNA input. Custom primers and probes are used for deep sequencing of sDNA treated with bisulfite to determine the methylation rate of DNA in each sample.
To quantify the level of methylation, we use the Probe Ex Taq (Probe qPCR) (TAKARA, RR390A, Kota Osaka, Japan) kit, and according to the manufacturer’s instructions, perform methylation-specific quantitative PCR (qMSP) detection on samples treated with bisulfite. According to the operating procedure of the Probe Ex Taq kit, the cycle threshold (CT value) is calculated by pre-determining the cutoff value for each amplification curve. Each batch of PCR reactions is performed with three controls, ACTB as an internal control, methylated cgl3096260 and cg12587766 as positive controls, and unmethylated cgl3096260 and cg12587766 as negative controls. Target gene capture, bisulfite treatment, and PCR amplification will be rerun using the second aliquot samples from the samples. Primers and probes are designed based on the specific sequences of CpG sites, with ACTB as the reference gene, and a two-step PCR amplification program is established according to the standard operating procedure. The PCR reaction instrument is AB 7500qPCR (Applied Biosystems, Foster City, CA, USA). The two-step PCR amplification cycle is as follows: 95 °C pre-denaturation for 30 s; 95 °C for 5 s, 60 °C for 30 s, repeated for 40 cycles. Detailed information on the primer and probe sequences of CpG sites and reference genes, molecular cloning vector plasmids, real-time fluorescence qPCR reaction conditions, and plasmid gradient dilution can be found in the supplementary materials and methods.
Determination of detection limit of methylation markers and inclusion of queues in the study
Synthetic plasmids with different template concentrations are diluted from plasmids constructed by Nanjing Kingsray Biotechnology Co., Ltd. with candidate CpG site sequences, and added to the aforementioned reaction system. Then, the detection limit of the methylation biomarker test is determined. Finally, it is determined that the Ct value of the reference ACTB methylation expression level should be less than 36, indicating that there is sufficient DNA for analysis, and samples with a Ct value higher than 36 are considered unqualified. For the methylation biomarkers cg13096260 and cg12587766, a Ct value less than 38 indicates a positive result, while a Ct value greater than 38 or not detected indicates a negative result.
In the study, a total of 878 patients participated from two medical centers, with 758 participants finally included in the study, including 62 participants with interfering diseases. Unfortunately, 64 patients were excluded due to unsuccessful collection of fecal samples or failure to obtain complete clinical case staging information, 29 participants were excluded due to insufficient reference genes, and 27 participants were excluded due to unqualified samples. In addition, DNA methylation detection was performed on the feces of 62 patients with interfering diseases (fecal samples included 48 cases of gastric malignant tumors, 3 cases of GIST, 8 cases of benign diseases, 1 case of gastric hyperplastic polyp, and 2 cases of liver malignant tumors). Among the 756 participants included in the analysis, 344 had CRC, 79 had AA, 273 had NED, and 62 had other interfering diseases. Supplementary File 1: Tables S2, S3: The main baseline characteristics of the samples are listed.
Data analysis
If the sample data follow a normal distribution, t-tests and chi-square tests are used, otherwise rank-sum tests are used. Single or multiple biomarkers are used to establish single-target and dual-target diagnostic logistic regression models based on the glm function. The random forest model in machine learning algorithms is used to establish a random forest diagnostic model. The ROC curve is used to calculate the AUC value, 95% confidence interval, specificity, and sensitivity, to evaluate the diagnostic performance of candidate methylation sites and guide the selection of cutoff points. All statistical analyses are performed using SPSS 26.0, R 4.1.1, and GraphPad Prism 8 software. The criteria for determining statistical significance are *P < 0.05, **P < 0.01, ***P < 0.0001, ns indicates no statistical significance.
Results
Screening of tissue-specific methylation markers and comprehensive analysis of DNA methylation expression
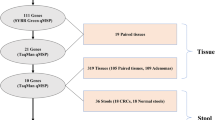
The screening process for tissue-specific CRC methylation marker sites is shown in Fig. 1. According to the screening process, two CpG sites that are highly specifically expressed in CRC and have low or no expression in normal tissues and other tumor tissues were identified through step-by-step screening. Among them, cg13096260 is located in the promoter region of SDC2, and cg12587766 is located on the LIFR gene.

Workflow for screening tissue-specific colorectal cancer methylation markers
Based on the analysis results from the TCGA database (Fig. 2A), the methylation level of cg13096260 located in the promoter region of the SDC2 gene is significantly higher in colorectal cancer (COAD, READ), diffuse large B cell lymphoma (DLBC), and stomach adenocarcinoma (STAD) tissues compared to normal tissues. Similarly, the methylation level of cg12587766 in the LIFR gene (Fig. 2B) is significantly higher in colorectal cancer (COAD, READ) and diffuse large B cell lymphoma (DLBC) tissues than in normal tissues. Subsequently, we analyzed the methylation sequencing data from 432 colorectal cancer and normal tissues in the TCGA database, comparing the DNA methylation β-values of cg13096260 and cg12587766 between normal and colorectal cancer tissues (Fig. 3: A, B). The results showed that the methylation levels of these two candidate CpG sites are significantly higher in colorectal cancer tissues compared to normal tissues.

Analysis of β-values for cg13096260 (A) and cg12587766 (B) across 33 common tumor tissues in the TCGA database; hierarchical clustering heatmaps of cg13096260 (C) and cg12587766 (D) between healthy controls and colorectal cancer patients; E negative correlation between methylation level of cg13096260 and transcriptional expression of the SDC2 gene; F negative correlation between methylation level of cg12587766 and transcriptional expression of the LIFR gene; G relationship between methylation level of cg13096260 and survival in colorectal cancer patients; H relationship between methylation level of cg12587766 and survival in colorectal cancer patients

Analysis of methylation levels of cg13096260 and cg12587766. The β values of cg13096260 (A) and cg12587766 (B) in the sequencing of colorectal cancer tissue samples in the TCGA database. The β values of DNA methylation of cg13096260 (C) and cg12587766 (D) in CRC and paired normal tissues were detected by pyrosequencing. E–F: methylation levels of cg13096260 and cg12587766 in fecal samples from NED, AA, and CRC patients collected by two medical centers
Moreover, differential analysis of cg13096260 and cg12587766 (Fig. 2C, D) revealed significant differences in methylation levels between colorectal cancer (CRC) patients and non-colorectal cancer (NED) patients. By analyzing the methylation β-values of cg13096260 and cg12587766 (Fig. 3A, B), we found that the methylation levels of these two CpG sites are significantly higher in colorectal cancer tissues than in normal tissues (P < 0.001).
Functional analysis of cg13096260 and cg12587766 (Fig. 2E and F) showed that cg13096260 is negatively correlated with the transcriptional expression level of SDC2 (P < 0.001), and cg12587766 is negatively correlated with the transcriptional expression level of LIFR (P < 0.001). Compared to normal colon tissues and other types of cancer, cg13096260 and cg12587766 exhibit specific high methylation modifications in colorectal cancer tissues, with corresponding downregulation in gene expression levels. Additionally, using survival data from colorectal cancer patients in the TCGA database, patients were divided into high and low methylation groups based on cutpoint values. Survival analysis (Fig. 2G and H) revealed that the methylation levels of these two sites are associated with the prognosis of colorectal cancer patients; higher methylation levels correlate with poorer prognosis.
Verification of DNA methylation status in CRC tissues using pyrosequencing
The DNA methylation status of the aforementioned two candidate genes in 6 cases of CRC and paired normal tissues was detected using pyrosequencing. The box plot of the average β values of the methylation status of these two genes is shown in Fig. 3C, D. The average differences in methylation levels of cgl3096260 and cg12587766 in cancer tissues and paired normal tissues are Δβ = 36.46 and Δβ = 19.372, respectively. The two genes show significantly different higher methylation (P < 0.05). This is consistent with the conclusions obtained through database annlyses, the methylation status of the screened genes can significantly distinguish between primary colorectal cancer and normal tissues. Hence, these two genes are incorporated into further validation studies.
Establishment of CRC diagnostic prediction model based on tissue-specific methylation markers in stools
The methylation levels of cgl3096260 and cg12587766 in 344 CRC, 79 AA, 273 NED, and 62 interfering disease fecal samples were detected by qMSP. It was found that the methylation level of cg13096260 in the NED group was significantly lower than that in the AA and CRC groups, and the methylation level in the AA group was significantly lower than that in the CRC group (Fig. 3E). It was found that the methylation level of Cg12587766 in the NED group was significantly lower than that in the CRC group, and the methylation level in the AA group was also significantly lower than that in the CRC group, but there was no significant difference between the AA group and the NED group. This may be related to the extremely low detection rate of cg12587766 in precancerous lesions and NED (5/273) patients (Fig. 3F).
To clarify whether methylation markers that perform well in tissues can be reproduced in fecal samples and have equally good diagnostic capabilities. We randomly divided 598 fecal samples into a training set and a validation set at a ratio of 7:3 (Fig. 4). The training set includes fecal samples from 200 cases of CRC, 54 cases of AA, and 165 cases of NED. The validation set includes fecal samples from 86 CRC patients, 25 AA patients, and 68 NED patients. This training set is used to evaluate the predictive ability of the two candidate CpG sites for the disease and to construct a joint diagnostic model. We constructed single-target models for cg13096260 and cg12587766, and a dual-target joint diagnostic model for cg13096260 and cg12587766. The cutoff value is established using the validation set to determine the clinical significance and verify the diagnostic efficacy of the two sites.

Workflow for constructing a diagnostic model using sDNA methylation markers
Establishment of logistic regression diagnostic model
Based on the Ct values of the two candidate CpG sites in the training set and their corresponding groups and the aforementioned sample threshold, the ROC curves of single target and dual target of different groups are obtained and the area under the curve (AUC) is calculated. The detection results are used to construct a single-target logistic regression diagnostic model (Fig. 5: A–C) and a dual-target logistic regression (LR combine) diagnostic model (Fig. 6: A–C). In the validation set, the aforementioned established models are used to construct ROC curves for different groups and calculate the AUC values of the corresponding groups. The area under the ROC curve (AUC) for the methylated cg13096260 and cg12587766 in the validation set was observed to be similar to that in the training set. The false-positive rates (FPR) for the SDC2 and LIFR genes were approximately 0.0203 and 0.0041, respectively. This suggests that these two candidate CpG sites have a high discriminatory ability for the disease and a low false-positive rate, indicating potential for large-scale screening (Fig. 5: D–F).It is determined that these two methylation markers will be used for further analysis and testing.

ROC curves and AUC of cg13096260 and cg12587766 in the fecal sample training set, predictive ability of AA and CRC (A), CRC (B), AA (C). ROC curves and AUC of cg13096260 and cg12587766 in the fecal sample validation set, predictive ability of AA and CRC (D), CRC (E), AA (F)

The predictive ability of the dual-target combined diagnostic (LR combine) model of cg13096260 and cg12587766 in the fecal sample training set for AA and CRC (A), CRC (B), AA (C). The predictive ability of the dual-target combined diagnostic model of cg13096260 and cg12587766 in the fecal sample training set within the random forest model (RF combine) for AA and CRC (A), CRC (B), AA (C). The predictive ability of the dual-target combined diagnostic (LR combine) model of cg13096260 and cg12587766 in the fecal sample validation set for AA and CRC (D), CRC (E), AA (F). The predictive ability of the dual-target combined diagnostic model of cg13096260 and cg12587766 in the fecal sample validation set within the random forest model (RF combine) for AA and CRC (D), CRC (E), AA (F)
Validation of the dual-target logistic regression diagnostic model
In the validation set, the ROC curve of the dual target was constructed through the Ct values of the two targets and the AUC value was calculated. Compared with the single-target model established by Cg13096260 and Cg12587766, the area under the ROC curve (AUC) of the dual target has improved to varying degrees. Measured by the area under the ROC curve (AUC), the dual-target model’s ability to distinguish between colorectal cancer, adenoma, and NED queues is significantly higher than that of the single-target model. Additionally, the specificity of the dual target for the CRC + AA cohort is 98.5% (CI 95.7–100%), and the sensitivity is 82.0% (CI 74.8%-89.1%). The specificity for CRC is 98.2% (CI 96.1–100%), and the sensitivity is 88.4% (CI 84.0–92.9%). For AA, the specificity is 98.5% (CI 95.7–100%), and the sensitivity is 75.9% (CI 64.5–87.3%). The false-positive rates for the combined model of cg13096260 and cg12587766 were 0.018 in the training set and 0.15 in the dual-target validation set. This demonstrates extremely high specificity and relatively high sensitivity, as well as very low false-positive rates. Therefore, dual-target DNA methylation detection can more effectively detect clinically typical lesions compared to single-target models.
Specificity verification in other gastrointestinal tumors and benign diseases
To further evaluate the specificity of the methylation sites in the study, a total of 62 patients with other interfering diseases were included in this study, and the methylation levels of the screening sites were tested. The negative rate of cg 13,096,260 in other gastrointestinal tumors and benign diseases is 84.91% (45/53) and 100% (9/9), respectively, with an overall negative rate of 87.10%. The negative rate of Cg12587766 in other gastrointestinal tumors and benign diseases is 100% (53/53) and 100% (9/9), respectively, with an overall negative rate of 100%. The negative rate of other gastrointestinal tumors and benign diseases in the combined diagnostic model is 84.91% (45/53) and 100% (9/9), respectively, with an overall negative rate of 87.10%. Based on the detection situation of interfering diseases, the selected methylation sites exhibit extremely high specificity.
Establishment of machine learning diagnostic prediction model
Machine learning is a subfield of artificial intelligence that uses statistical methods to optimize models for specific tasks without predefining all rules or parameters. Thus, machine learning models may outperform regression models.
In this study, we assessed the influence of machine learning and logistic diagnostic models on the diagnostic performance of dual targets. Based on the Ct values of the two candidate CpG sites in the training set and their corresponding groups, the ROC curve of the dual targets was constructed using the random forest algorithm, and the area under the curve (AUC) value was calculated. The dual-target combination random forest (RF combine) diagnostic model was built using the detection results (Fig. 6: A–F). Compared to the logistic regression model, the random forest diagnostic model showed improved diagnostic efficiency. The sensitivity of the RF combine model for the CRC + AA cohort is 91.3% (CI 87.8%-94.8%), and the specificity is 95.8% (CI 92.7–98.8%). For CRC, the sensitivity is 95.5% (CI 92.6–98.4%), and the specificity is 95.8% (CI 92.7–98.8%). For AA, the sensitivity is 81.5% (CI 71.1–91.8%), and the specificity is 97.6% (CI 95.2–99.9%). Additionally, in the random forest model, the false-positive rate of the dual target in the training set for AA + CRC versus Normal mixed samples is 0.042, and in the validation set, it is 0.015. For AA versus Normal samples, the false-positive rate in the training set is 0.024, and in the validation set, it is 0.029. Compared to single-target and conventional regression models, the random forest model has higher sensitivity and specificity and a lower false-positive rate. This suggests that machine learning algorithms may have advantages in establishing diagnostic prediction models and merit further exploration and development.
Verification of the logistic diagnostic model and random forest diagnostic model with dual-center data
To further assess the stability and performance of the established model, fecal samples were collected from 72 patients with colorectal cancer who visited the Gastrointestinal Surgery Department of Sichuan Provincial People’s Hospital from June 2022 to March 2023, 28 patients with other gastrointestinal tumors (7 out of 36 patients were excluded due to unqualified samples, 1 was excluded due to lack of clinical data), and 41 patients without disease (1 out of 42 patients was excluded due to unqualified samples) for validation. The baseline table of the subjects can be seen in Additional file: Table S2.
Using the established models, ROC curves were constructed for different groups, and AUC values were calculated for each group. It was found that the area under the ROC curve (AUC) of methylated cg13096260 and cg12587766 in the validation set was similar to that in the training set (cg13096260 AUC = 0.8482, 95% CI 0.7864 − 0.9099; cg12587766 AUC = 0.6967, 95% CI 0.6349 − 0.7585), indicating that our targets maintain good disease differentiation ability across centers (Fig. 7A). In the LR combine model, the AUC in the central validation set was 0.8613 (95% CI 0.8029 − 0.9196), showing good performance(Fig. 7B). The AUC of the RF combine diagnostic model was 0.8664 (95% CI 0.799 − 0.9337) (Fig. 7C). The false-positive rate for cg13096260 was 0.025, and for cg12587766 it was 0.00. This indicates that the selected markers exhibit good differentiation ability and low false-positive rates in other centers.

Validation of the single-target and dual-target logistic regression (LR) models and random forest models at another center, including the single-target LR diagnostic model (A), the dual-target LR combined diagnostic model (B), and the dual-target RF combined diagnostic model(C)
To further assess the specificity of the methylation sites under study at another center, fecal samples from 28 patients with other gastrointestinal tumors but without colorectal tumors were incorporated into this research, and the methylation levels of the selected sites were examined. The negativity rate of cg 13,096,260 in other gastrointestinal tumors is 85.8% (4/28). The negativity rate of cg12587766 in other gastrointestinal tumors and benign diseases is 100% (28/28). The negativity rate of the combined diagnostic model for other gastrointestinal tumors is 87.30% (4/28). This indicates that the selected methylation sites still have extremely strong specificity in the samples of other centers.
Discussion
Timely removal of late-stage adenomas can effectively reduce the incidence of colorectal cancer, and timely diagnosis of early-stage colorectal cancer followed by radical surgery at the resectable stage can significantly reduce its mortality. Commonly used tools for colorectal cancer screening include the fecal occult blood test (gFOBT), fecal immunochemical test (FIT), colonoscopy, and fecal DNA testing, among others. Large-scale randomized controlled trials and observational studies have found that gFOBT, FIT, and colonoscopy can reduce the mortality rate of colorectal cancer patients [23]. However, the low specificity and sensitivity of gFOBT and FIT, misdiagnosis and missed diagnosis may occur. Simultaneously, colonoscopy, being an invasive examination, has low compliance, requires complex preoperative preparation, and may carry risks such as discomfort, bleeding, and perforation, making it difficult to expand the scope of screening.
The occurrence of colorectal cancer is closely related to genetic and epigenetic changes [24]. The DNA of tumor cells shed in feces contains cancer-specific genetic and epigenetic changes, so it can be used as a marker for noninvasive diagnosis of colorectal cancer. Differential methylation analysis of colorectal cancer tissues and normal tissues to screen for potential sDNA methylation markers is a commonly used method. The frequency of SDC2 promoter methylation in colorectal cancer tumor tissues is extremely high, which is a marker with great potential [25]. Yoon Dae Han and others evaluated the methylation level of SDC2 in the free DNA of 585 patients to assess the application ability of SDC2 in the early detection of colorectal cancer. They found that the sensitivity and specificity of SDC2 are both over 90.2% [26]. Imperiale and others developed the first multi-target free DNA methylation detection kit Cologuard™ using fecal DNA methylation detection and fecal immunochemical detection. Its sensitivity to colorectal cancer is 92.3%, its sensitivity to late precancerous lesions is 42.4%, and its specificity is 86.6% [17]. Although we have found many biomarkers with high sensitivity, it is still uncertain whether these markers have tissue specificity. Methylation markers used for colorectal cancer detection in the past may be interfered with by other cancers and lack tissue specificity. For example, SEPT9, which is currently approved for clinical detection of colorectal cancer, is expressed in multiple cancers such as colorectal cancer, cervical cancer [27], and gastric cancer [28].If methylation markers lacking tissue specificity are applied to the clinic, they have high sensitivity, which may lead to erroneous further examinations or misdiagnoses. This contradicts our principle of expecting precise screening through sDNA; therefore, we believe that cancer screening should have tissue specificity.
To select colorectal cancer methylation biomarkers with tissue specificity, we utilized a large public methylation database to assess the methylation levels of the chosen markers in common tumor tissues and normal tissues, to ascertain whether these markers are specifically found in colorectal cancer. Through this screening method, we can maximize the assurance that the selected markers have a high degree of tissue specificity for the detection of colorectal cancer and its precancerous lesions. Finally, we screened out two methylation sites with high tissue specificity, cg13096260 located in the SDC2 gene and cg12587766 located in the LIFR gene.
In this study, we first used pyrosequencing to validate the DNA methylation levels of specific CpG sites of the biomarkers screened from the database in actual clinical samples. We then measured the sDNA methylation levels of cg13096260, cg12587766, and the reference gene in the feces of colorectal cancer (CRC), adenoma (AA), and no pathological evidence disease (NED) patients using qMSP to test their tissue specificity and sensitivity in real clinical samples. We found that the cg13096260 marker showed high sensitivity and fairly high specificity in diagnosing colorectal cancer and its precancerous lesions. In contrast, the biomarker cg12587766 performs moderately in diagnosing precancerous lesions but shows an extremely low detection rate among patients with precancerous lesions and those without pathological evidence, indicating exceptionally high specificity. Therefore, the cg12587766 marker may help improve the specificity of dual target and diagnosis in some cases. Ultimately, the outcomes of our model prediction also substantiated this perspective.
In this study, we separately utilized cg13096260 and cg12587766 to construct single-target and dual-target combined diagnostic models. We found that both models could precisely differentiate between colorectal cancer patients and normal individuals, exhibiting extremely high specificity and sensitivity, and maintained good stability in external data validation. Compared with FIT testing, our study's sensitivity was 95.5% versus 73.8%, and specificity was 95.8% versus 94.9% [17]. Compared with the first clinically used SEPT9 assay, sensitivity was 95.5% versus 90.2%, and specificity was 95.8% versus 90.2% [26]. Compared with the established multi-target detection assay Cologuard™, sensitivity for colorectal cancer was 95.5% versus 92.3%, and specificity was 95.8% versus 86.6% [17]. Additionally, the dual-target model in our study showed a sensitivity of 81.5% and a specificity of 97.6% for adenomas. Compared with current related models, the methylation markers selected in our study may have greater value in diagnosing colorectal cancer and precancerous lesions and even possess the potential for early detection of asymptomatic colorectal cancer patients or adenomas.
A significant highlight of this research is that we constructed a dual-target diagnostic model utilizing the random forest algorithm, and we tested its performance in the training queue and validation queue. The ROC curve demonstrates that, in comparison with the logistic diagnostic model, the random forest diagnostic model possesses stronger predictive capability and stronger differentiation potential. Hence, it is more probable to become a potent tool for predicting precancerous changes and colorectal cancer, which will assist us in more accurately identifying patients who require active examination and treatment. Nonetheless, clinical prediction models constructed by machine learning algorithms necessitate the analysis of a vast amount of data, and if the datasets utilized for development and usage differ, data drift might occur, potentially leading to the model misidentifying and generating incorrect diagnoses. Consequently, we employed an independent dataset from another center to validate the stability of the random forest model. Even though the model still exhibits excellent performance on the new dataset, to thoroughly assess its reliability in clinical decision-making, further verification is required on a larger scale and data from different centers.
To our knowledge, although a few previous studies reported that the two genes corresponding to the CpG sites found in our study may have the potential to diagnose colorectal cancer, they did not validate the actual diagnostic ability of these genes in actual clinical samples or dual-target models [29, 30]. Cho discovered, utilizing quantitative methylation-specific PCR (qMSP), that the methylation frequency in the LIFR promoter region in colon cancer tissues is significantly higher than in normal colon tissues and mucosal tissues of non-cancer patients [31]. Dapeng Li noted the current absence of colorectal cancer markers with tissue specificity and discovered and validated that cg18174928, cg12602374, and cg11841722 situated in the LIFR promoter region possess tissue specificity, and thus they might serve as noninvasive sDNA markers for diagnosing colorectal cancer [32]. Coincidentally, another research found that cg13096260 could potentially act as a screening and early diagnostic marker for colorectal cancer and precancerous changes, but the study did not substantiate its tissue specificity, nor was it verified utilizing an external dataset.
In our study, we used the whole-genome methylation data of various cancers in the bioinformatics database to find markers with tissue specificity, which provides a possible research path for finding methylation markers with colorectal cancer tissue specificity. Our research demonstrates innovation in the following points. Firstly, we discovered two CpG sites with tissue-specific methylation patterns. In a substantial number of real clinical specimens, we assessed the sensitivity and specificity of these two sites in sDNA testing and constructed various predictive models. Secondly, considering that other types of gastrointestinal tumors might interfere with the sites we screened, we also incorporated other types of gastrointestinal tumors and benign diseases to evaluate the robustness of our screened sites and dual-target models. Thirdly, we introduced an external dataset to validate the diagnostic capability of the sites we screened and the models we constructed. Lastly, our research also found that machine learning algorithms might be a more promising method of modeling, which is vital for us to create a more advantageous clinical predictive model.
Naturally, our research also has some limitations. First, our sample database contains only 33 common types of tumors, meaning similar targets present in other rare types of tumors or benign diseases might not be included. Secondly, although the dual-target model using the random forest algorithm outperformed the logistic regression model in classification performance, it may produce more false-positive results in certain cases. Therefore, further testing and evaluation in more centers are needed to ensure the model's stability. Additionally, most of the subjects participated in the screening due to symptoms like gastrointestinal bleeding, diarrhea, and changes in bowel habits, so the detection sensitivity for asymptomatic screening subjects might be lower than our reported data. Lastly, all subjects have the same genetic background, and future studies may need to consider racial differences.
Conclusions
This study conducted a detailed assessment of the methylation patterns of cg13096260 and cg12587766, validating the efficacy of these two sites, both individually and in combination, for diagnosing colorectal cancer and precancerous changes. Additionally, through the evaluation of different modeling methods, we found that machine learning algorithms exhibit superior performance. Therefore, the results of this study support the subsequent development of screening kits and the implementation of large-scale randomized clinical trials to validate its clinical applicability and explore the use of different modeling methods to improve diagnostic efficacy. However, we also note that multi-target models may lead to more false-positive results while increasing sensitivity and specificity.
Availability of data and materials
The gene testing data utilized in this study will be available in the supplementary materials of the article, for the portions that can be publicly shared. Data containing sensitive personal information are protected by privacy laws and will not be disclosed without specific authorization. For access to the study data, please contact QinXing Cao via email (Email: 2,423,806,408@qq.com).
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Zheng R, Zhang S, Zeng H, et al. Cancer incidence and mortality in China, 2016. J Natl Cancer Center. 2022;2(1):1–9.
Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin May-Jun. 2023;73(3):233–54.
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394(10207):1467–80.
Berger BM, Ahlquist DA. Stool DNA screening for colorectal neoplasia: biological and technical basis for high detection rates. Pathology. 2012;44(2):80–8.
Zorzi M, Fedeli U, Schievano E, et al. Impact on colorectal cancer mortality of screening programmes based on the faecal immunochemical test. Gut. 2015;64(5):784–90.
Brenner H, Stock C, Hoffmeister M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: systematic review and meta-analysis of randomised controlled trials and observational studies. BMJ. 2014;348:g2467.
Schreuders EH, Ruco A, Rabeneck L, et al. Colorectal cancer screening: a global overview of existing programmes. Gut. 2015;64(10):1637–49.
Hong SN. Genetic and epigenetic alterations of colorectal cancer. Intest Res. 2018;16(3):327–37.
Grady WM, Yu M, Markowitz SD. Epigenetic alterations in the gastrointestinal tract: current and emerging use for biomarkers of cancer. Gastroenterology. 2021;160(3):690–709.
Heiss JA, Brenner H. Epigenome-wide discovery and evaluation of leukocyte DNA methylation markers for the detection of colorectal cancer in a screening setting. Clin Epigenetics. 2017;9:24.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–59.
Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–34.
Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–86.
Oster B, Thorsen K, Lamy P, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer. 2011;129(12):2855–66.
Ahlquist DA, Zou H, Domanico M, et al. Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology. 2012;142(2):248–256; quiz e225–246.
Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;370(14):1287–97.
Guo H, Cheng Y, Martinka M, McElwee K. High LIFr expression stimulates melanoma cell migration and is associated with unfavorable prognosis in melanoma. Oncotarget. 2015;6(28):25484–98.
Liu SC, Tsang NM, Chiang WC, et al. Leukemia inhibitory factor promotes nasopharyngeal carcinoma progression and radioresistance. J Clin Invest. 2013;123(12):5269–83.
Wu HX, Cheng X, Jing XQ, et al. LIFR promotes tumor angiogenesis by up-regulating IL-8 levels in colorectal cancer. Biochim Biophys Acta Mol Basis Dis. 2018;1864(9 Pt):2769–84.
Song P, Li Y, Wang F, et al. Genome-wide screening for differentially methylated long noncoding RNAs identifies LIFR-AS1 as an epigenetically regulated lncRNA that inhibits the progression of colorectal cancer. Clin Epigenetics. 2022;14(1):138.
Chen M, Zhao H. Next-generation sequencing in liquid biopsy: cancer screening and early detection. Hum Genom. 2019;13(1):34.
Ladabaum U, Dominitz JA, Kahi C, Schoen RE. Strategies for colorectal cancer screening. Gastroenterology. 2020;158(2):418–32.
Jung G, Hernandez-Illan E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol. 2020;17(2):111–30.
Oh T, Kim N, Moon Y, et al. Genome-wide identification and validation of a novel methylation biomarker, SDC2, for blood-based detection of colorectal cancer. J Mol Diagn. 2013;15(4):498–507.
Han YD, Oh TJ, Chung TH, et al. Early detection of colorectal cancer based on presence of methylated syndecan-2 (SDC2) in stool DNA. Clin Epigenetics. 2019;11(1):51.
Jiao X, Zhang S, Jiao J, et al. Promoter methylation of SEPT9 as a potential biomarker for early detection of cervical cancer and its overexpression predicts radioresistance. Clin Epigenetics. 2019;11(1):120.
Zhao L, Li M, Zhang S, Liu Y. Plasma-methylated SEPT9 for the noninvasive diagnosis of gastric cancer. J Clin Med. 2022;11(21):6399.
Shen Y, Wang D, Yuan T, et al. Novel DNA methylation biomarkers in stool and blood for early detection of colorectal cancer and precancerous lesions. Clin Epigenetics. 2023;15(1):26.
Wang X, Wang D, Zhang H, Feng M, Wu X. Genome-wide analysis of DNA methylation identifies two CpG sites for the early screening of colorectal cancer. Epigenomics. 2020;12(1):37–52.
Cho YG, Chang X, Park IS, et al. Promoter methylation of leukemia inhibitory factor receptor gene in colorectal carcinoma. Int J Oncol. 2011;39(2):337–44.
Li D, Zhang L, Fu J, et al. Discovery and validation of tissue-specific DNA methylation as noninvasive diagnostic markers for colorectal cancer. Clin Epigenetics. 2022;14(1):102.
Funding
The study was funded by the Key R&D project of the Science and Technology Department of Sichuan Province (Project No. 2023YFS0185), Key R&D project of the Science and Technology Department of Sichuan Province (Project No. 2023YFS0197), Sichuan Provincial Department of Science and Technology, Sichuan Province Natural Science Foundation (Project No. 2022NSFSC1541), University of Electronic Science and Technology of China, and the "Medical-Engineering Cross Joint Fund" of University of Electronic Science and Technology of China—Sichuan Provincial People’s Hospital (Project No. ZYGX2021YGLH022), Key Research and Development Support Program of Chengdu Science and Technology Bureau (2024-YF05-01714-SN).
Author information
Authors and Affiliations
Contributions
QC, ZD, and NY contributed to conducting the experiments, software, validation, formal analysis, writing the original draft, visualization, data management, and supervision. LY, XY, HL, SY were responsible for resources, sample collection and processing, and clinical data organization. JZ, HX, QL, and XZ conducted experiments, database analysis, sample collection and processing, and resource management. MP and MZ were involved in conceptualization, investigation, project administration, and funding acquisition. All authors reviewed the manuscript and provided their final approval for submission.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This research has received ethical approval from the Ethics Committee of the Sichuan Academy of Medical Sciences and Sichuan Provincial People's Hospital (Affiliated Hospital of University of Electronic Science and Technology) under the ethical approval number: Ethical Application Review (Research) 2022 No. 306. Written informed consent was provided by each participant before any sample collection.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, Q., Dan, Z., Hou, N. et al. Discovery and validation of colorectal cancer tissue-specific methylation markers: a dual-center retrospective cohort study. Clin Epigenet 16, 122 (2024). https://doi.org/10.1186/s13148-024-01735-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01735-6