
Abstract
Background
Atrial natriuretic peptide (ANP), one of the main members of the natriuretic peptides system, has been associated with hypertension and related complications, but the underlying molecular mechanisms are not very clear. Here, we aimed to examine whether DNA methylation, a molecular modification to the genome, of the natriuretic peptide A gene (NPPA), the coding gene of ANP, was associated with hypertension.
Methods
Peripheral blood DNA methylation of NPPA promoter was quantified by target bisulfite sequencing in 2498 community members (mean aged 53 years, 38% men) as a discovery sample and 1771 independent participants (mean aged 62 years, 54% men) as a replication sample. In both samples, we conducted a single CpG association analysis, followed by a gene-based association analysis, to examine the association between NPPA promoter methylation and hypertension, adjusting for age, sex, education level, cigarette smoking, alcohol consumption, obesity, fasting glucose, and lipids. Multiple testing was controlled by the false discovery rate approach.
Results
Of the 9 CpG loci assayed, hypermethylation at 5 CpGs (CpG1, CpG3, CpG6, CpG8, and CpG9) was significantly associated with a lower odds of prevalent hypertension in the discovery sample, and one CpG methylation (CpG1 located at Chr1:11908353) was successfully replicated in the replication sample (OR = 0.82, 95%CI 0.74–0.91, q = 0.002) after adjusting for covariates and multiple testing. The gene-based analysis found that DNA methylation of the 9 CpGs at NPPA promoter as a whole was significantly associated with blood pressure and prevalent hypertension in both samples (all P < 0.05).
Conclusions
DNA methylation levels at NPPA promoter were decreased in Chinese adults with hypertension. Aberrant DNA methylation of the NPPA gene may participate in the mechanisms of hypertension.
Similar content being viewed by others


Background
Hypertension, a leading modifiable risk factor for cardiovascular morbidity and mortality, affects two thirds of the population and accounts for the largest disease burden in the world [1]. Although many risk factors of hypertension have been established [2,3,4], decades of control efforts made through interventions against these factors did not effectively improve the prevention and management of this global epidemic which is still increasingly prevalent, indicating that some unknown mechanisms may exist. In China, nearly 245 million adults suffered from hypertension but only 15% of them were under control of blood pressure [5]. Fundamentally, blood pressure elevation reflects increased cardiac output or peripheral resistance. As an important cardiac endocrine regulatory system for the body in response to external environmental stimuli, the natriuretic peptides system plays a critical role in maintaining salt-water balance and blood pressure through natriuresis, diuresis, and vasodilation in response to cardiac output overload [6, 7]. As one of the main members of the natriuretic peptides system, atrial natriuretic peptide (ANP) may play a vital role in the development of hypertension. Indeed, the relationship between ANP and hypertension has been demonstrated by substantial evidence. For example, transgenic mice overexpressing ANP [8, 9] exhibited reduced blood pressure and mice with functional disruptions in ANP had an elevated blood pressure [10, 11] compared with their wild-type littermates. In humans, the circulating level of ANP has been associated with hypertension [12] and related vascular complications, e.g., atherosclerosis [13], heart failure [14], and stroke [15]. Polymorphisms of its coding gene—natriuretic peptide A (NPPA)—have also been associated with the susceptibility to hypertension [16,17,18], stroke [15, 19], myocardial infarction [20], and coronary artery disease [21, 22]. Notably, the association of rs5068 located at the NPPA gene with hypertension reached a genome-wide significance with a P value of 1 × 10−8 [18]. The recombinant ANP carperitide has been used in clinical practice for the management of acute decompensated heart failure [23, 24] but could cause some unfavorable effects, e.g., severe hypotension [24,25,26] and in-hospital death [27,28,29]. A better understanding of the molecular mechanisms of the blood pressure-regulating effect of ANP may help its drug development and improvement. As an interface between the fixed genome and dynamic environment, DNA methylation of gene promoters has been found to play a critical role in the regulation of transcriptional activity and gene expression [30]. Therefore, we hypothesized that aberrant methylation of the NPPA promoter may affect its function and subsequent ANP synthesis and excretion, thereby participating in the pathogenesis of hypertension, but this has not been studied in humans. Here, we aimed to examine the association between NPPA promoter methylation and hypertension in two independent samples of Chinese adults.
Results
Clinical characteristics of study participants
As described in Fig. 1, a total of 2498 participants (mean aged 53 years, 38% men) in the Gusu cohort, a community-based study of cardiovascular disease and its risk factors in middle-aged and elderly Chinese adults [31], were included in the current study as the discovery sample and 1771 participants (mean aged 62 years, 54% men) in a case-control study designed to identify epigenetic markers of ischemic stroke were included as the replication sample. Of them, 1109 (44.39%) and 995 (56.18%) participants have been diagnosed with hypertension in the two samples. Their clinical characteristics were shown in Table 1. In both study samples, hypertensive participants, as expected, were more likely to be older and have more metabolic risk factors including obesity, fasting glucose, and blood lipids than those without hypertension (all P < 0.05).

A flowchart illustrating the selection of study participants
Results in the discovery sample
As illustrated in Fig. 2, DNA methylation levels at a total of 9 CpG loci in the promoter region of the NPPA gene were assayed by target bisulfite sequencing using genomic DNA isolated from peripheral blood mononuclear cells. DNA methylation levels at all of the 9 CpG loci assayed were significantly lower in participants with hypertension than those without after correction for multiple testing by adjusting for the total number of CpG loci tested using the false discovery rate (FDR) approach (all q < 0.05, Table 2). Of them, hypomethylation of 3 CpGs was significantly associated with either SBP or DBP, independent of conventional risk factors including age, sex, education level, cigarette smoking, alcohol consumption, body mass index (BMI), fasting glucose, lipids, and antihypertensive medications. After correction for multiple testing, hypermethylation at only 1 CpG (CpG1 located at Chr1:11908353) was still significantly associated with a lower level of SBP (β = − 0.96, q < 0.05). Hypermethylation at 4 additional CpGs (CpG3 located at Chr1:11908299, CpG6 located at Chr1:11908178, CpG8 located at Chr1:11908165, and CpG 9 located at Chr1:11908142) was also significantly associated with a lower odds of prevalent hypertension after adjusting for covariates and multiple testing (all q < 0.05). As expected, DNA methylation levels at the neighboring CpG sites were highly correlated (Supplementary Figure S1), suggesting a co-methylation pattern in the promoter region of the NPPA gene. Therefore, we examined whether DNA methylation at NPPA promoter, rather than a single CpG site, was associated with hypertension. The results showed that the average methylation level of all CpGs was significantly associated with a lower risk of prevalent hypertension (OR = 0.88, 95%CI 0.81–0.97, P = 0.008). Also, the weighted truncated product method (wTPM), an approach combines the effect of each single CpG methylation, found that DNA methylation of the 9 CpGs at the NPPA promoter as a whole was significantly associated with SBP, DBP, and prevalent hypertension (all P < 0.05, Table 3).

A schematic illustration of the targeted sequence and primers for targeted bisulfite sequencing. Red represents the CpG loci assayed in the NPPA gene promoter (− 540 to about − 277 bp from TSS). TSS, transcriptional start site
Results in the replication sample
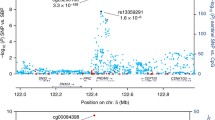
A similar phenomenon of the association between NPPA promoter methylation and hypertension was observed in the replication sample. Almost all CpG methylation levels seemed to be lower in participants with hypertension than those without, but only 3 (CpG1, CpG3, CpG6) survived after the correction of multiple testing (all q < 0.05, Table 2). Hypomethylation of 8 CpGs including CpG1 located at Chr1:11908353 (β = − 2.22 for SBP and β = − 1.34 for DBP) were significantly associated with either SBP or DBP after adjusting for covariates and multiple testing (all q < 0.05, Table 4). Consistent with findings in the discovery sample, hypermethylation at CpG1 was also significantly associated with a lower odds of prevalent hypertension after adjusting for covariates and multiple testing (OR = 0.82, 95%CI 0.74–0.91, q = 0.002, Fig. 3). The gene-based analysis found that DNA methylation of the 9 CpGs at NPPA promoter as a whole was significantly associated with SBP, DBP, and prevalent hypertension (all P < 0.05).

The effects of DNA methylation at CpG1 located at Chr1:11908353 on blood pressure and hypertension in the discovery and replication samples. SBP, systolic blood pressure; DBP, diastolic blood pressure; HBP, hypertension
Results of secondary analysis
The meta-analysis did not identify more CpG sites whose methylation levels were associated with blood pressure or hypertension (Supplementary Figure S2). Bioinformatics analysis showed that the NPPA gene participated in 7 KEGG pathways and 30 biological processes, most of which have been considered involved in the regulation of blood pressure (Supplementary Table S1). Gene network analysis found similar results that the NPPA gene may play a critical role in the pathogenesis of hypertension through varied functions, e.g., circulatory system process, regulation of blood pressure, and blood circulation (Supplementary Figure S3).
Discussion
Leveraging two independent samples of Chinese adults, we found for the first time that DNA methylation levels of NPPA promoter were lower in participants with hypertension than those without. Hypermethylation at the promoter region of the NPPA gene was associated with a lower risk of prevalent hypertension, independent of behavior and metabolic factors. These results may suggest a potential role of aberrant NPPA promoter methylation in the molecular mechanisms of hypertension.
As one of the main components of the natriuretic peptides system, ANP has been widely considered a potential drug candidate for the cardiovascular system due to its biological functions of natriuresis, diuresis, vasorelaxation, and inhibition of renin and aldosterone secretion [32]. The recombinant ANP carperitide has been used in clinical practice for the management of acute decompensated heart failure [23, 24] but could cause some unfavorable effects, e.g., severe hypotension [24,25,26] and in-hospital death [27,28,29]. A better understanding of the molecular mechanisms underlying the association between ANP and hypertension may help its drug development and improvement. In line with our study, the role of its coding gene—NPPA—in blood pressure regulation has been suggested by previous studies [16, 17, 33,34,35]. For example, a meta-analysis including 4068 individuals revealed that the NPPA gene rs5065 polymorphism might contribute to the occurrence of hypertension [33]. A recent GWAS identified a hypertension-related SNP rs3753584 located at the NPPA gene which was associated with a higher circulating ANP level [35]. These findings suggest that factors regulating the function of the NPPA gene may conserve the potential to be the molecular mechanisms emerging to be studied that underneath the association between ANP and hypertension. As a modifiable molecular modification to the genome without changes in the genes’ sequence, DNA methylation may affect gene function and repress transcription by altering promoter DNA accessibility and blocking the binding of transcription activating proteins [36]. We, therefore, hypothesized that the NPPA promoter methylation may be a potential molecular modification that regulates ANP expression/excretion and participate in the pathogenesis of hypertension. Indeed, this epigenetic regulation of ANP expression has been observed by integrative analyses on DNA methylomes and transcriptomes data in humans [37]. Some hypertension-related methylation markers have been found in animals and humans. The hypomethylation of angiotensin II receptor type 1b might lead to overexpression of the gene in the adrenal glands and the development of hypertension in adult rats [38, 39]. In humans, aberrant methylation of angiotensin II receptor type 1 promoter [40] and angiotensin I converting enzyme [41] was associated with hypertension. An epigenome-wide association study including 17,010 individuals identified many CpG sites and genes in association with blood pressure, but none was related to the NPPA gene [42]. No study, to the best of our knowledge, has examined the role of DNA methylation at the NPPA gene in hypertension. Leveraging an unselected population in the Gusu cohort, we are the first to examine the association between NPPA promoter methylation and hypertension and provide initial evidence for the potential role of NPPA promoter methylation in the pathogenesis of hypertension. We found that DNA methylation level of NPPA promoter was more likely to be lower in participants with hypertension than those without. Hypomethylation of the NPPA gene was suggested to be associated with an upregulation of ANP transcription [37]. In line with our study, many studies have found that patients with hypertension had higher levels of ANP than those without and ANP levels showed a progressive elevation with the severity of hypertension [12, 43,44,45]. Although we did not have data on circulating ANP in our study, together with prior studies, we believed that DNA methylation of the NPPA gene undoubtedly participated in the molecular mechanisms of hypertension. The causality of DNA methylation of the NPPA gene in hypertension development is still unclear and warranted to be further studied.
In this study, we found that the contribution of an individual CpG methylation to blood pressure or hypertension was in general small (mostly < 5%), and statistically most CpG sites could not withstand multiple testing correction. Such a small effect size may not be detected by conventional statistical methods. Their combined effects may be large enough to be useful for risk prediction. Therefore, we tested the joint associations of multiple CpG methylation with blood pressure or hypertension and found significant joint associations in both sample, even though most single CpG associations did not reach a statistical significance level. Our results may unravel a molecular mechanism that NPPA promoter methylation may participate in the pathology of hypertension and suggest that simultaneously testing the joint effects of multiple CpG sites is a powerful approach in epigenetic analysis for complicated diseases, e.g., hypertension.
The strengths of our study include the independent replication of the association between NPPA promoter methylation and hypertension; comprehensive measurement and adjustment of confounding factors including lifestyles, metabolic factors, and medical history; and application of the wTPM to test the joint association of multiple CpG methylation at NPPA promoter with hypertension. Some limitations of our study should also be acknowledged. First, the cross-sectional study design precludes causal inference. It is still unclear whether aberrant methylation at NPPA promoter is a risk factor, consequence, or just an accompanying phenomenon of hypertension. Second, as in most observational studies, residual confounding is of concern. Although our results remained significant after controlling for known risk factors of hypertension and were successfully replicated in an independent sample, some unmeasured confounding effects may exist. Third, our findings were derived from Chinese adults only whose cardiovascular health profiles could be different from other populations with different ethnic backgrounds. Thus, the generalizability of our results to other ethnic populations is uncertain. Fourth, the participants in the replication sample come from a case-control study designed to identify epigenetic markers of ischemic stroke and seem to be older than those in the discovery sample. Selection bias may therefore exist in the replication sample and may overestimate the magnitude of the association between NPPA promoter methylation and hypertension. Fifth, we did not have data on circulating ANP levels in our study. Whether and to what extent NPPA promoter methylation accounts for the molecular mechanisms underlying the association between ANP and hypertension still needs more investigation, although hypomethylation of the NPPA gene was found to be associated with an upregulation of the transcripts of ANP [37]. Finally, given that DNA methylation is tissue- and cell-type specific, it is unclear whether or to what extent the results derived from peripheral blood could reflect methylation changes in the target organs of hypertension, e.g., cardiac and artery. However, accumulating evidence indicated that epimutations may not be limited to the affected tissue but could also be detected in peripheral blood [46,47,48].
Conclusions
In conclusion, we demonstrate that hypermethylation at NPPA promoter is associated with a lower level of blood pressure and a lower risk of having hypertension in Chinese adults. Our results suggest a potential role of NPPA promoter methylation in the molecular mechanisms of hypertension.
Methods
Study participants
As described in Fig. 1, the current study included 2498 participants as the discovery sample and 1771 participants as the replication sample.
Discovery sample
The Gusu cohort is a community-based prospective longitudinal study of cardiovascular disease and its risk factors in middle-aged and elderly Chinese adults. The study design, methods, and laboratory techniques have been described previously [31]. In brief, a total of 2706 community members over 30 years were included and completed the baseline examination conducted in 2010. All participants had not been diagnosed with cardiovascular or chronic kidney disease. The protocols were approved by the Soochow University Ethics Committee. Written informed consent was obtained from all study participants. After excluding participants lacking DNA samples (n = 208), 2498 participants were included in the final analysis as the discovery sample.
Replication sample
An independent sample of 1771 participants (including 853 ischemic stroke patients and 918 age- and sex-matched healthy controls) were selected from China Antihypertensive Trial in Acute Ischemic Stroke (CATIS) [49] and Metabolic Syndrome and the Multi-metabolic Disorders Study [50], respectively, based on the availability of DNA specimens. The primary objective of this study was to identify new epigenetic markers of ischemic stroke.
Quantification of the NPPA promoter methylation
In both samples, DNA methylation levels in the promoter region of the NPPA gene were quantified by target bisulfite sequencing as previously described [48]. using genomic DNA isolated from peripheral blood mononuclear cells. In brief, based on the genomic coordinates of the NPPA promoter in Genome Reference Consortium Human Build 37 (GRCh37), we carefully designed the primers to detect the maximum CpG loci within the CpG islands. The targeted sequence (Chr1: 11908117–11908380, reverse strand, relative to TSS: − 540 bp to − 277 bp) was illustrated in Fig. 2. Following primer validation, genomic DNA was bisulfite-treated using the EZ DNA Methylation-Gold Kit (Zymo Research, Inc., CA, USA) according to the manufacturer’s protocol, which converts unmethylated cytosine into uracil and leaves methylated cytosine unchanged. The treated samples were amplified, barcoded, and sequenced by Illumina Hiseq 2000 (Illumina, Inc., CA, USA) using the paired-end sequencing protocol according to the manufacturer’s guidelines. Methylation level at each CpG dinucleotides was calculated as the percentage of the methylated alleles over the sum of methylated and unmethylated alleles. For quality control, the samples with bisulfite conversion rate < 98% and the cytosine sites with average coverage less than 20× were filtered out. DNA methylation levels were finally quantified at 9 CpG loci in the NPPA promoter.
Measurement of blood pressure
Blood pressure was measured three times by trained staff using a standard mercury sphygmomanometer and a cuff of appropriate size according to a standard protocol [51], after the participants had been resting for at least 5 min in a relaxed, sitting position. The first and fifth Korotkoff sounds were recorded as systolic blood pressure (SBP) and diastolic blood pressure (DBP), respectively. The mean of the three measurements was used in statistical analyses. According to the Chinese guidelines for the management of hypertension, participants with an SBP ≥ 140 mmHg and/or a DBP ≥ 90 mmHg or under antihypertensive treatment in the last 2 weeks were diagnosed with hypertension [52].
Assessment of risk factors of hypertension
Demographic data including age, sex, and education level were obtained by questionnaires administered by trained staff. Cigarette smoking was classified as current smoking or not. Current smoking was defined as having smoked at least 100 cigarettes in the entire life, having smoked cigarettes regularly, and smoking currently. Alcohol consumption was classified as current drinkers or not. Current drinkers were those who had consumed any alcohol during the past year. Bodyweight (kg) and height (cm) were measured when participants wore light clothes and no shoes by trained staff. BMI was calculated by dividing weight in kilograms by the square of height in meters (kg/m2). Fasting glucose, blood lipids including total cholesterol, triglycerides, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) were measured by standard laboratory methods [31].
Statistical analysis
Baseline characteristics and NPPA promoter methylation levels were presented and compared between participants with and without hypertension using Student’s t test and Chi-square test as appropriate. We examined and replicated the cross-sectional association between DNA methylation levels of 9 CpG sites in the NPPA promoter region and prevalent hypertension in two independent samples using identical regression models. Multiple testing was controlled by adjusting for the total number of CpG loci tested using the FDR approach, and an FDR-adjusted P value (i.e., q value) of less than 0.05 was considered statistically significant. All analyses were conducted using R version 3.6.1.
Single CpG association analysis
To examine the association between each single CpG methylation and blood pressure, we constructed a linear regression model in which blood pressure (SBP and DBP, respectively) was the dependent variable and DNA methylation at each CpG locus was the independent variable, adjusting for age, sex, education level, cigarette smoking, alcohol consumption, BMI, fasting glucose, LDL-C, HDL-C, and antihypertensive medications (y/n). To facilitate data interpretation, we similarly constructed a logistic regression model with prevalent hypertension (y/n) as the dependent variable to further examine the association between NPPA promoter methylation and odds of hypertension, adjusting for age, sex, education level, cigarette smoking, alcohol consumption, BMI, fasting glucose, LDL-C, and HDL-C.
Gene-based association analysis
To examine whether DNA methylation of multiple CpG sites was jointly associated with hypertension, we treated the average methylation level of multiple CpG sites as a substitute for the methylation level of the targeted region and similarly examined its associations with blood pressure and prevalent hypertension. We also employed the wTPM as previously described [53], based on the results of a single CpG association analysis. This method combines P values of all CpGs that reaches a preselected threshold (e.g., raw P < 0.1 in this study). The regression coefficient of each individual CpG methylation was included as weights in the wTPM statistic.
Secondary analysis
To identify more potential CpG sites, we conducted a meta-analysis using the summary data from the discovery and replication samples. The random-effects model was applied to provide more conservative estimated effects. Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted for the NPPA gene by DAVID [54]. The regulatory network involving the NPPA gene was also constructed by GeneMANIA (http://genemania.org/).
Availability of data and materials
The datasets used during the current study are available from the corresponding author on a reasonable request.
Abbreviations
- ANP:
-
Atrial natriuretic peptide
- NPPA :
-
Natriuretic peptide A
- GRCh37:
-
Genome Reference Consortium Human Build 37
- SBP:
-
Systolic blood pressure
- DBP:
-
Diastolic blood pressure
- BMI:
-
Body mass index
- HDL-C:
-
High-density lipoprotein cholesterol
- LDL-C:
-
Low-density lipoprotein cholesterol
- FDR:
-
False discovery rate
- wTPM:
-
Weighted truncated product method
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Moser M. Update on the management of hypertension: recent clinical trials and the JNC 7. J Clin Hypertension (Greenwich, Conn). 2004;6(10 Suppl 2):4–13.
Gu D, Gupta A, Muntner P, Hu S, Duan X, Chen J, et al. Prevalence of cardiovascular disease risk factor clustering among the adult population of China: results from the International Collaborative Study of Cardiovascular Disease in Asia (InterAsia). Circulation. 2005;112(5):658–65.
Li Y, Feng X, Zhang M, Zhou M, Wang N, Wang L. Clustering of cardiovascular behavioral risk factors and blood pressure among people diagnosed with hypertension: a nationally representative survey in China. Scientific reports. 2016;6:27627.
Ying CQ, Fu SB, Xu Q, Tong WJ, Fang MW, Wu ZL, et al. Multiple risk factor clustering and risk of hypertension in the Mongolian ethnic population of China. Biomedical and environmental sciences : BES. 2007;20(5):381–5.
Wang Z, Chen Z, Zhang L, Wang X, Hao G, Zhang Z, et al. Status of hypertension in China: results from the China Hypertension Survey, 2012-2015. Circulation. 2018;137(22):2344–56.
Zois NE, Bartels ED, Hunter I, Kousholt BS, Olsen LH, Goetze JP. Natriuretic peptides in cardiometabolic regulation and disease. Nature reviews Cardiology. 2014;11(7):403–12.
Volpe M, Rubattu S, Burnett J Jr. Natriuretic peptides in cardiovascular diseases: current use and perspectives. Eur Heart J. 2014;35(7):419–25.
Steinhelper ME, Cochrane KL, Field LJ. Hypotension in transgenic mice expressing atrial natriuretic factor fusion genes. Hypertension (Dallas, Tex : 1979). 1990;16(3):301–7.
Ogawa Y, Itoh H, Tamura N, Suga S, Yoshimasa T, Uehira M, et al. Molecular cloning of the complementary DNA and gene that encode mouse brain natriuretic peptide and generation of transgenic mice that overexpress the brain natriuretic peptide gene. The Journal of clinical investigation. 1994;93(5):1911–21.
John SW, Veress AT, Honrath U, Chong CK, Peng L, Smithies O, et al. Blood pressure and fluid-electrolyte balance in mice with reduced or absent ANP. The American journal of physiology. 1996;271(1 Pt 2):R109–14.
Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14730–5.
Macheret F, Heublein D, Costello-Boerrigter LC, Boerrigter G, McKie P, Bellavia D, et al. Human hypertension is characterized by a lack of activation of the antihypertensive cardiac hormones ANP and BNP. Journal of the American College of Cardiology. 2012;60(16):1558–65.
Casco VH, Veinot JP, Kuroski de Bold ML, Masters RG, Stevenson MM, de Bold AJ. Natriuretic peptide system gene expression in human coronary arteries. J Histochem Cytochem. 2002;50(6):799–809.
Maisel AS, Duran JM, Wettersten N. Natriuretic peptides in heart failure: atrial and B-type natriuretic peptides. Heart failure clinics. 2018;14(1):13–25.
Xing DG, Zhang DY, Wang ZF, Ding DL, Wang J, Wang YJ. Correlations of ANP genetic polymorphisms and serum levels with ischemic stroke risk: a meta-analysis. Genetic testing and molecular biomarkers. 2014;18(5):349–56.
Conen D, Glynn RJ, Buring JE, Ridker PM, Zee RY. Natriuretic peptide precursor a gene polymorphisms and risk of blood pressure progression and incident hypertension. Hypertension (Dallas, Tex : 1979). 2007;50(6):1114–9.
Conen D, Cheng S, Steiner LL, Buring JE, Ridker PM, Zee RY. Association of 77 polymorphisms in 52 candidate genes with blood pressure progression and incident hypertension: the Women's Genome Health Study. J Hypertens. 2009;27(3):476–83.
Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nature genetics. 2009;41(3):348–53.
Rubattu S, Stanzione R, Di Angelantonio E, Zanda B, Evangelista A, Tarasi D, et al. Atrial natriuretic peptide gene polymorphisms and risk of ischemic stroke in humans. Stroke. 2004;35(4):814–8.
Rubattu S, Sciarretta S, Marchitti S, Bianchi F, Forte M, Volpe M. The T2238C human atrial natriuretic peptide molecular variant and the risk of vardiovascular diseases. Int J Mol Sci. 2018;19(2):540.
Ziaee S, Kalayinia S, Boroumand MA, Pourgholi L, Cheraghi S, Anvari MS, et al. Association between the atrial natriuretic peptide rs5065 gene polymorphism and the presence and severity of coronary artery disease in an Iranian population. Coronary artery disease. 2014;25(3):242–6.
Barbato E, Bartunek J, Mangiacapra F, Sciarretta S, Stanzione R, Delrue L, et al. Influence of rs5065 atrial natriuretic peptide gene variant on coronary artery disease. Journal of the American College of Cardiology. 2012;59(20):1763–70.
Ogiso M, Isogai T, Okabe Y, Ito K, Tsuji M, Tanaka H. Effect of carperitide on in-hospital mortality of patients admitted for heart failure: propensity score analyses. Heart and vessels. 2017;32(8):916–25.
Nomura F, Kurobe N, Mori Y, Hikita A, Kawai M, Suwa M, et al. Multicenter prospective investigation on efficacy and safety of carperitide as a first-line drug for acute heart failure syndrome with preserved blood pressure: COMPASS: Carperitide Effects Observed Through Monitoring Dyspnea in Acute Decompensated Heart Failure Study. Circulation journal : official journal of the Japanese Circulation Society. 2008;72(11):1777–86.
Suwa M, Seino Y, Nomachi Y, Matsuki S, Funahashi K. Multicenter prospective investigation on efficacy and safety of carperitide for acute heart failure in the 'real world' of therapy. Circulation journal : official journal of the Japanese Circulation Society. 2005;69(3):283–90.
Hattori H, Minami Y, Mizuno M, Yumino D, Hoshi H, Arashi H, et al. Differences in hemodynamic responses between intravenous carperitide and nicorandil in patients with acute heart failure syndromes. Heart and vessels. 2013;28(3):345–51.
Mizuno A, Iguchi H, Sawada Y, Hurley M, Nomura H, Hayashi K, et al. The impact of carperitide usage on the cost of hospitalization and outcome in patients with acute heart failure: High value care vs. low value care campaign in Japan. Int J Cardiology. 2017;241:243–8.
Kobayashi D, Yamaguchi N, Takahashi O, Deshpande GA, Fukui T. Human atrial natriuretic peptide treatment for acute heart failure: a systematic review of efficacy and mortality. The Canadian journal of cardiology. 2012;28(1):102–9.
Nagai T, Iwakami N, Nakai M, Nishimura K, Sumita Y, Mizuno A, et al. Effect of intravenous carperitide versus nitrates as first-line vasodilators on in-hospital outcomes in hospitalized patients with acute heart failure: Insight from a nationwide claim-based database. International journal of cardiology. 2019;280:104–9.
Zhu H, Wu LF, Mo XB, Lu X, Tang H, Zhu XW, et al. Rheumatoid arthritis-associated DNA methylation sites in peripheral blood mononuclear cells. Ann Rheumatic Dis. 2019;78(1):36–42.
Peng H, Zhang Q, Cai X, Liu Y, Ding J, Tian H, et al. Association between high serum soluble corin and hypertension: a cross-sectional study in a general population of China. American journal of hypertension. 2015;28(9):1141–9.
Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handbook of experimental pharmacology. 2009;191:341–66.
Niu W. The relationship between natriuretic peptide precursor a gene T2238C polymorphism and hypertension: a meta-analysis. Int J Hypertens. 2011;2011:653698.
Arora P, Wu C, Khan AM, Bloch DB, Davis-Dusenbery BN, Ghorbani A, et al. Atrial natriuretic peptide is negatively regulated by microRNA-425. The Journal of clinical investigation. 2013;123(8):3378–82.
Salo PP, Havulinna AS, Tukiainen T, Raitakari O, Lehtimäki T, Kähönen M, et al. Genome-wide association study implicates atrial natriuretic peptide rather than B-type natriuretic peptide in the regulation of blood pressure in the general population. Circ Cardiovasc Gene. 2017;10(6):e001713.
Marx A, Kahan T, Simon I. Integrative analysis of methylome and transcriptome reveals the importance of unmethylated CpGs in non-CpG island gene activation. BioMed Res Int. 2013;2013:785731.
Meder B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Frese K, Lai A, et al. Epigenome-wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation. 2017;136(16):1528–44.
Bogdarina I, Haase A, Langley-Evans S, Clark AJ. Glucocorticoid effects on the programming of AT1b angiotensin receptor gene methylation and expression in the rat. PloS One. 2010;5(2):e9237.
Bogdarina IG, King PJ, Clark AJ. Characterization of the angiotensin (AT1b) receptor promoter and its regulation by glucocorticoids. J Mol Endocrinol. 2009;43(2):73–80.
Fan R, Mao S, Zhong F, Gong M, Yin F, Hao L, et al. Association of AGTR1 promoter methylation levels with essential hypertension risk: a matched case-control study. Cytogenet Genome Res. 2015;147(2-3):95–102.
Rangel M, dos Santos JC, Ortiz PH, Hirata M, Jasiulionis MG, Araujo RC, et al. Modification of epigenetic patterns in low birth weight children: importance of hypomethylation of the ACE gene promoter. PloS One. 2014;9(8):e106138.
Richard MA, Huan T, Ligthart S, Gondalia R, Jhun MA, Brody JA, et al. DNA Methylation analysis identifies loci for blood pressure regulation. Am J Hum Genet. 2017;101(6):888–902.
Sagnella GA, Markandu ND, Shore AC, MacGregor GA. Raised circulating levels of atrial natriuretic peptides in essential hypertension. Lancet (London, England). 1986;1(8474):179–81.
Arendt RM, Gerbes AL, Ritter D, Stangl E, Bach P, Zähringer J. Atrial natriuretic factor in plasma of patients with arterial hypertension, heart failure or cirrhosis of the liver. J Hypertens Suppl. 1986;4(2):S131–5.
Ekholm D, Edvinsson L, Ekman R, Thulin T. Plasma atrial natriuretic peptide (ANP) in relation to blood pressure in severe hypertension. Journal of internal medicine. 1992;231(3):281–5.
Menke A, Binder EB. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin Neurosci. 2014;16(3):395–404.
Uddin M, Koenen KC, Aiello AE, Wildman DE. de los Santos R, Galea S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychol Med. 2011;41(5):997–1007.
Pu W, Wang C, Chen S, Zhao D, Zhou Y, Ma Y, et al. Targeted bisulfite sequencing identified a panel of DNA methylation-based biomarkers for esophageal squamous cell carcinoma (ESCC). Clinical epigenetics. 2017;9:129.
He J, Zhang Y, Xu T, Zhao Q, Wang D, Chen CS, et al. Effects of immediate blood pressure reduction on death and major disability in patients with acute ischemic stroke: the CATIS randomized clinical trial. Jama. 2014;311(5):479–89.
Zhang L, Guo Z, Wu M, Hu X, Xu Y, Zhou Z. Interaction of smoking and metabolic syndrome on cardiovascular risk in a Chinese cohort. International journal of cardiology. 2013;167(1):250–3.
Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. Jama. 2003;289(19):2560–72.
Liu LS. 2010 Chinese guidelines for the management of hypertension. Zhonghua Xin Xue Guan Bing Za Zhi. 2011;39(7):579–615.
Zaykin DV, Zhivotovsky LA, Westfall PH, Weir BS. Truncated product method for combining P-values. Genetic epidemiology. 2002;22(2):170–85.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57.
Acknowledgements
We are deeply appreciative of the participants in this study and thank all staffs for their support and assistance. Especially, we thank the clinical staff at all participating hospitals for their support and contribution to this project. Without their contribution, this research would not have been possible.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81903384 and 81872690), the Natural Science Foundation of Jiangsu Province (No. BK20180841), the Suzhou Municipal Science and Technology Bureau (No. SS201853), and a Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JL and JZ performed the statistical data analysis and drafted the manuscript. HP developed the concept of the study design and contributed to draft the manuscript. LR, SM, BS, and YH obtained the clinical data and critically reviewed the manuscript. JY, RZ, and MZ contributed to the interpretation of the results. All authors contributed to draft the final versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study protocols were approved by the institutional review boards at Soochow University in China and Tulane University in the USA, as well as the ethical committees of all participating hospitals. Written informed consent was obtained from all study participants or their immediate family members.
Consent for publication
All authors have read and approved the submission and the manuscript has not been published and is not being considered for publication elsewhere in whole or part, in any language.
Competing interests
None of the authors have financial associations that might pose a conflict of interest in connection with the submitted article. The datasets used during the current study are available from the corresponding author on reasonable request.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table S1.
Biologic processes for the NPPA gene identified by Gene Ontology (GO). Supplementary Figure S1. The Pearson correlation matrix among DNA methylation levels of all CpG loci assayed in NPPA promoter in the discovery sample. Supplementary Figure S2. Results of the meta-analysis. Supplementary Figure S3. Gene net-work involving the NPPA gene.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, J., Zhu, J., Ren, L. et al. Association between NPPA promoter methylation and hypertension: results from Gusu cohort and replication in an independent sample. Clin Epigenet 12, 133 (2020). https://doi.org/10.1186/s13148-020-00927-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-020-00927-0