
Abstract
It is accepted that cancer stem cells (CSCs) are key to the occurrence, progression, drug resistance, and recurrence of bladder cancer (BLCA). Here, we aimed to characterize the landscapes of CSCs and investigate the biological and clinical signatures based on a prognostic model constructed by genes associated with CSCs. The malignant epithelial cells were discovered and sorted into six clusters through single cell analysis. C2 was identified as the CSCs. The signaling involved in the interactions between C2, cancer-associated fibroblasts (CAFs), and immune cells mainly consisted of MK, THBS, ANGPTL, VISFATIN, JAM, and ncWNT pathways. The CSC-like prognostic index (CSCLPI) constructed by the random survival forest was a reliable risk factor for BLCA and had a stable and powerful effect on predicting the overall survival of patients with BLCA. The level of CAFs was higher among patients with higher CSCLPI scores, suggesting that CAFs play a significant role in regulating biological characteristics. The CSCLPI-developed survival prediction nomogram has the potential to be applied clinically to predict the 1-, 2-, 3-, and 5-year overall survival of patients with BLCA. The CSCLPI can be used for prognostic prediction and drug treatment evaluation in the clinic.
Similar content being viewed by others

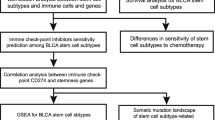

Introduction
Bladder cancer (BLCA) is a prevalent type of malignant tumor in the urogenital system, resulting in approximately 550, 000 new cases and 200, 000 deaths annually [1]. The incidence rate is affected by gender differences, with males having an incidence rate about three times that of females [1]. There are different molecular subtypes and pathogenic pathways for BLCA, and the disease can be classified as non-muscle invasive (NMIBC) (70% of cases of first detected tumors) or muscle invasive (MIBC) (30%) in clinic. BLCA consists of 75% urothelial carcinoma and 25% histopathology labeled “variant” (e.g. squamous differentiation, micropapillary) [2]. Identification of variant histological subtypes accurately is crucial for risk stratification, as these variants exhibit aggressive biological behavior and relate to advanced disease at presentation. In the majority of cases, radical cystectomy along with pelvic lymph node dissection is the chosen treatment method. The value of this classification lies in its ability to reflect the response to drug therapy (e.g. neoadjuvant chemotherapy, immunotherapy). Importantly, NMIBC and MIBC are where the clinical treatment differences and prognosis differences are most apparent. NMIBC is usually treated with a combination of transurethral resection of a bladder tumor (TURBT) and a risk stratified approach to adjuvant intravesical therapy (IVe). For MIBC, surgery was the main focus of a comprehensive treatment plan assisted by immune checkpoint inhibitors, targeted treatments, and antibody-drug conjugates [3]. BLCA is a disease that remains challenging to treat due to its heterogeneous nature with specific genetic and epigenetic characteristics.
Stem cells are known for their pluripotency and self-renewal potential. It is believed that a complex tumor microenvironment contains cancer stem cells (CSCs), including BLCA. Bladder CSCs were firstly discovered in 2009 using markers to distinguish them from normal basal cells [4]. The subsequent research also backed up this viewpoint and identified several markers for stem cells [5, 6]. Shen et al. have found that BLCA has a high expression of SOX4 that can promote stem cell properties [5]. Moreover, its expression was linked to advanced cancer stages and a poor prognosis. Zhu et al. have discovered that B-cell-specific Moloney murine leukemia virus insertion site 1 (Bmi1) is crucial for maintaining stemness properties and tumorigenicity of human bladder CSC-like cells [6]. These results provide a novel insight into understanding the occurrence, progression, drug resistance, and recurrence of BLCA. However, the complexity in the tumor microenvironment prevents further observation of the CSCs’ characterization and crosstalk between other cell populations. The milestone technology of single-cell sequencing has become a reality for depicting the landscape of the tumor microenvironment. Wang et al. identified distinct cell subpopulations and discovered that the cancer stem-cell subpopulation becomes enriched during bladder cancer recurrence with an increase in EZH2 expression [7]. Ma et al. identified the subtypes of CSCs and mesenchymal stem cells in the tumor microenvironment of BLCA using single-cell sequencing and explored their interactions based on a ligand-target network [8]. This study seeks to reveal the characterization of the stem cell landscape utilizing single-cell sequencing, and attempts to develop and validate a stem cells-like model using machine learning and to investigate its clinical and biological signatures.
Methods and materials
Acquiring scRNA-seq and bulk RNA-seq data
The Gene Expression Omnibus (GEO) database was accessed to download the BLCA scRNA-seq dataset by using accession numbers: GSE135337 [9], including 7 tumor samples and 1 normal sample.
BLCA bulk RNA-seq datasets were procured from TCGA and GEO databases with the following accession number: TCGA-BLCA, GSE13507 [10], GSE32894 [11], GSE48075 [12], and GSE48276 [13]. The TCGA-BLCA dataset had 412 tumor samples and 19 normal samples that were utilized as the training cohort. The corresponding clinical information contains overall survival (OS) time, survival status, age, gender, pathologic stage, and TNM stage. The matrix files for the four bulk RNA-seq transcriptomes were normalized, and they were regarded as the external validation cohorts.
Analyzing BLCA scRNA-seq data
The initial step in preparing scRNA-seq data was to screen single cells by ensuring that each gene was expressed in at least 5 cells and each cell had at least 300 genes expressed. Cells were filtered using the Seurat based on the number of genes detected per cell (200–7500) and the percentage of mitochondrial genes expressed (< 10%) [14]. The gene expression matrix was devoid of mitochondrial genes and ribosomal genes. At last, there were 36,680 cells remaining. The Seurat manual outlined approaches to integration, normalization, dimension reduction, and clustering [14]. With default parameters, the Seurat package’s FindVariableFeatures function identified the top 3000 genes that are highly variable. After Z-score normalization, PCA was carried out on genes with high variability. Significant principal components (PCs) were utilized to perform uniform manifold approximation and projection (UMAP) dimension reduction. Clusters were identified using the FindClusters function with a resolution of 0.8. Using the FindAllMarkers function, markers genes for each cluster were identified by comparison to one cluster with others with logFC = 0.25, minpct = 0.25, and adjusted p-value = 0.05. Afterwards, markers that have been reported in literature were used to determine and annotate different cell clusters (myeloid cells: PTPRC, CD14, CD68; T cells: PTPRC, CD3D, CD3E; epithelial cells: EPCAM, KRT18; stromal cells: ACTA2, MYL9, COL1A1, PDPN, VWF, and PECAM1). By using the CopyKAT R package, the CNV characteristics of the epithelial cell clusters were examined to discriminate malignant epithelial cells (ECs) from nonmalignant ECs [15]. To further identify malignant clusters, we analyzed the expression of BLCA markers, such as UPK3A, MTHFD2, UHRF1, FGFR3, ERBB2, BRCA1, CDKN2A, and BRAF [16,17,18,19,20,21,22,23], in normal clusters, ECs, and malignant ECs. Moreover, functional enrichment analysis was carried out on the genes that differed in expression between malignant ECs and ECs.
Pseudotime trajectory analysis for malignant ECs
A pseudotime trajectory was established using Monocle 2 that was based on the gene expression profiles of malignant ECs [24]. The malignant ECs were projected and ordered into a trajectory with different branches, and the cells within the same branch were believed to have the same state of differentiation. To identify genes with branch-dependent expression patterns, branched expression analysis modeling (BEAM) (qval < 1e-20) was applied. The identification of genes that are branch-dependent or state-specific by clustering genes at the branch point led to the investigation of the mechanisms behind the cell fate decision.
CytoTRACE
The CytoTRACE offers an innovative approach to measure cell differentiation capacity, which can lead to a significant improvement in predicting cell differentiation at the single-cell level [25]. It has been proven to perform better than current computational methods for stemness assessment in large-scale datasets. Each malignant EC is given a stemness score by the CytoTRACE algorithm after calculating the gene expression matrix. A higher CytoTRACE score is indicative of a higher stemness (lower differentiation) for malignant ECs.
Analyzing the regulatory network of malignant ECs
The activation of the transcription factor (TF) responsible for gene regulation in each malignant EC phenotype was determined by using decoupleR [26]. The tool is capable of inferring biological activities using a variety of methods, making it efficient, robust, and user-friendly for omics data analysis. Moreover, it offers wrappers that are easy to use to query the OmniPath meta-database. OmniPath offers a common syntax for various methods, types of omics datasets, and knowledge sources that can be used in multiple workflows to facilitate the exploration of different approaches.
Metabolic analysis of malignant ECs
We used scMetabolism (version 0.2.1) [27] to quantify metabolism activity at the single-cell resolution for analyzing metabolism activity of malignant ECs. Each cell had its metabolic score calculated using the sc.metabolism.Seurat function, with the type of metabolism being “KEGG”.
Functional enrichment analysis
The biological characteristics of genes that were differentially expressed were investigated through functional enrichment analysis using the “clusterProfiler” package, as well as an online website (Metascape: http://metascape.org/gp/index.html#/main/step1), which were used to perform KEGG pathway analysis and Gene Ontology (GO) analysis. Seurat’s FindMarkers function was used to identify genes that were differentially expressed in scRNA-seq data, while “EdgeR” package was used to obtain them in bulk RNA-seq data with the adjusted P-value < 0.05 and |log2 [FC]| >1.
Cell-cell communication analysis using CellChat
CellChat is a new program that uses scRNA-seq data to quantify the intercellular communication networks. The Cellchat package (version: 1.6.1) was used to predict the intercellular communication between CSCs subtype and stromal cells in BLCA, which was determined by analyzing ligand-receptor interactions. Briefly, we utilized CellChat to assess the most important signaling inputs and outputs among CSCs and stromal cells clusters via CellChatDB.human. The netVisual_circle function was utilized to demonstrate the strength or weakness of cell–cell communication networks. The netVisual_bubble function presents bubble plots of important ligand-receptor interactions.
Machine learning-based integrative approaches to generate CSCs-related genes
Univariate Cox regression was used initially to identify promising markers for prognosis among C2 cluster gene sets. The screening hub CSC-related genes was then carried out with 10 machine-learning algorithms and 99 combinations. The integrative algorithms comprised random survival forest (RSF), elastic network (Enet), Lasso, Ridge, stepwise Cox, CoxBoost, partial least squares regression for Cox (plsRcox), supervised principal components (SuperPC), generalized boosted regression modeling (GBM), and survival support vector machine (survival-SVM). The machine learning algorithms were followed as in a previous study [28]. The TCGA cohort was set as the training cohort, and the test cohort contained the GSE13507, GSE32894, GSE48075, and GSE48276 cohorts. Harrell’s concordance index (C-index) was determined in all cohorts. The one with the highest average C-index is considered the optimal model. A variable selection function was used by the optimal model to identify the hub CSCs-related genes.
Construction and validation of a cancer stem cell-like prognostic index (CSCLPI)
The genes associated with hub CSCs that were identified through the optimal model were incorporated into the multivariate Cox regression model to establish a cancer stem cell-like prognostic index (CSCLPI), by multiplying the expression values of genes by their coefficient in the model and then adding them together. The median CSCLPI value was used to divide the patients into high- and low-CSCLPI groups. Then, we performed KM survival analysis on the relevant hub CSCs genes with statistical significance and the CSCLPI to estimate their prognostic power. CSCLPI was also developed in the GSE13507, GSE32894, GSE48075, and GSE48276 cohorts. The median CSCLPI value of the TCGA cohort was used to divide patients in the GSE13507, GSE32894, GSE48075, and GSE48276 cohorts into high- and low-CSCLPI groups. KM survival analysis was also conducted on them. The ROC curve was used to analyze the performance of CSCLPI in predicting 1-, 2-, and 3-year overall survival of BLCA patients.
Analysis of gene mutation in CSCLPI subgroups
The discrepant gene mutation in CSCLPI subgroups was detected by conducting mutation analysis on BLCA samples. The major mutation genes and variant classifications in CSCLPI subgroups were a source of concern for us. The investigation involved examining the differences in mutated genes between two subgroups.
Evaluation of CSCLPI-based landscape of tumor microenvironment
The Immune score and Stromal score were evaluated with the use of the ESTIMATE algorithm [29] in the high CSCLPI and low CSCLPI groups. A single-sample gene set enrichment analysis (ssGSEA) algorithm was used to evaluate the abundance of immune cells and the score of immune-related activity or function [30]. Tumor immune dysfunction and exclusion (TIDE) [31] is an effective tool for assessing tumor immune escape and has the potential to predict patient responses to immune therapy. TIDE and EPIC [32] were applied to quantify the relative proportions of infiltrating immune cells and cancer-associated fibroblasts (CAFs).
Exploration of the characteristic differences in CSCLPI subgroups
The messenger RNA expression-based stemness index (mRNAsi) of each BLCA sample (ranges from zero to one) was calculated using one-class logistic regression machine-learning algorithm (OCLR) based on pluripotent stem cell samples, which was related to stem cell features and could be applied to predict cancer stemness [33]. After normalizing and filtering low-expression data, the “TCGAbiolinks” package was used to obtain mRNAsi of BLCA with gene information as “geneInfo”, method as “quantile” and stemSig as “SC_PCBC_stemSig”. The prognostic value of mRNAsi and the correlation between CSCLPI and mRNAsi were analyzed. GO and KEGG enrichment analyses were used to analyze differentially expressed genes between the CSCLPI subgroups to investigate biological signatures and potential regulatory pathways. The optimal category amount was determined by using consensus clustering analysis to classify BLCA samples based on the cancer-associated fibroblast-specific markers previously reported [34]. The samples of different fibroblast subtypes in the CSCLPI subgroups, as well as the expression of cancer-associated fibroblast-specific markers, were evaluated.
Drug sensitivity prediction
The “oncoPredict” R package was employed to evaluate drug sensitivity to conventional chemotherapy among high- and low-CSCLPI patients by comparing their half-maximum inhibitory concentration (IC50) [35].
Development and validation a nomogram
Clinical characteristics (including age, gender, pathologic_stage, pathologic_T, and pathologic_N) and CSCLPI were investigated in Univariate and multivariate Cox regression analyses to determine their impact on BLCA patients’ overall survival. A nomogram based on these factors was developed to predict the 1-, 2-, 3-, and 5-year prognosis. The C-index, calibration curve, and DCA curve were used to estimate the model’s accuracy. The heatmap displayed the association between screened CSCs and the clinical characteristics, CSCLPI and the fibroblast subtypes.
Statistical analysis
Statistical analyses and plots were carried out using R software. The continuous variables were compared by independent t-test or Wilcoxon’s rank-sum test. Univariate and multivariate prognostic analysis was conducted with the use of Cox regression models. Analysis of survival was conducted using the Kaplan-Meier method and log-rank test. The time-dependent ROC curve was plotted using the “timeROC” R package. The DCA curve was depicted using the “ggDCA” R package. The threshold for statistical significance was P < 0.05.
Results
Cell subtypes
The analysis process of the article was represented in Fig. 1. With stringent quality control metrics in place prior to pre-processing, we managed to obtain 36,680 high-quality cellular samples from 7 BLCA tissues (Supplementary Fig. 1A). The number of genes and the sequencing depth were found to have a strong positive correlation after normalization, with a Pearson correlation coefficient of 0.92 (Supplementary Fig. 1B). With a resolution of 0.8, 20 clusters were identified (Fig. 2A). The cell type annotation was based on the markers that have been reported in literature (Supplementary Fig. 1C). Thus, clusters 15, 17, and 19 were identified as immune cells; cluster 16 was identified as stromal cells; remaining clusters were identified as epithelial cells (Fig. 2B and C), which were consistent with previous classification results [9]. Based on single-cell transcriptome data from BLCA samples, without relying on normal cells as controls, 3 and 8 clusters (a total of 4962 cells) were identified as malignant epithelial cells (ECs) using CopyKAT (Fig. 2D, Supplementary Fig. 1D). Compared to epithelial cells and normal clusters, the 3 and 8 clusters showed high expression of BLCA markers (Supplementary Fig. 1E). The results of functional enrichment analysis for differentially expressed genes between malignant ECs and ECs demonstrated that some signaling pathways associated with carcinogenesis were activated, including VEGFA and TP53 signaling pathways (Supplementary Fig. 1F). These results supported that 3 and 8 clusters were cancerous clusters.

The flowchart of analysis

Identification of cell subtypes. (A) 20 clusters are identified at a resolution of 0.8. (B) Markers that identify different cells are expressed. We identify three major cell clusters: epithelial cells, stromal cells, and immune cells. (C) The UAMP plot displays the distribution of epithelial cells, stromal cells, and immune cells. (D) The results of CopyKAT for predicting malignant ECs. 3 and 8 clusters are considered malignant ECs.
Identification of stem cells cluster for BLCA
We drew 3 and 8 clusters of cells from the total cell population. They were subjected to the process of normalization, dimension reduction, and clustering once again. With a resolution of 0.25, 6 clusters were identified (Fig. 3A). The main differential genes of each cluster were shown in the Fig. 3B. GO and KEGG enrichment analysis for the 6 clusters showed that genes in cluster 1 (C1) were enriched in the signaling pathways that were related to chemical carcinogenesis- reactive oxygen species, glutathione metabolism et al.; genes in cluster 2 (C2) were enriched in the signaling pathways involved in pyrimidine metabolism, platinum drug resistance, cell cycle, DNA replication et al.; genes in cluster 3 (C3) were enriched in the signaling pathways implicated in mineral absorption, antigen processing and presentation et al.; genes in cluster 4 (C4) were enriched in the signaling pathways that were associated with leukocyte transendothelial migration, adhere junction, bladder cancer et al.; genes in cluster 5 (C5) were enriched in the signaling pathways that were connected to p53 signaling pathway activation, HIF − 1 signaling pathway activation, cellular senescence et al.; genes in cluster 6 (C6) were enriched in the signaling pathways that were related to antigen processing and presentation, Th1 and Th2 cell differentiation et al. (Fig. 3C, Supplementary Fig. 2A-2 F). The pseudotime trajectory analysis of malignant ECs was performed with Monocle2 to elucidate the developmental trajectory of the ECs (Fig. 3D) and to show that different clusters were at different stages of differentiation (Fig. 3E). Based on this finding, it can be inferred that C2 or C1 were the initial stages of differentiation in ECs. Furthermore, CytoTRACE revealed that C2 was the initiator in the predicted trajectory with higher differentiation potential, and C5 and C3 were the terminal states in the differentiation trajectory (Fig. 3F and G). Thus, C2 was identified as the cancer stem cells for BLCA for further analysis.

Cell differentiation trajectory. (A) 6 clusters of malignant ECs are identified with a resolution of 0.25. (B) Some of positively differential genes in each cluster. (C) Display the differential genes and KEGG pathway enrichment results of each cluster simultaneously. (D) A color-coded pseudotime plot depicts the differentiation trajectory of malignant ECs in BLCA. (E) Differentiation trajectory of malignant ECs in BLCA for sub-cell types. (F-G) CytoTRACE predicts the cell differentiation potential of malignant ECs in BLCA, and identified C2 as the cancer stem cell cluster
The characterization of the stem cell landscape of BLCA
Firstly, we investigated the activity of transcription factors (TFs) and the activation of main pathways, and discovered that in the C2 cluster, E2F1, E2F2, E2F3, E2F4, and TFAM were significantly activated (Fig. 4A, Supplementary Fig. 3A). PI3K and MAPK signaling pathways were involved in regulating the biological functions of the C2 cluster (Fig. 4B, Supplementary Fig. 3B). Secondly, through metabolic analysis, we were able to identify the main metabolic pathways in C2 cluster, including butanoate metabolism, citrate cycle (TCA cycle), fatty acid degradation and elongation et al. (Fig. 4C, Supplementary Fig. 3C), indicating changes in metabolism of CSCs. Thirdly, we drew 16 cluster (stromal cells) of cells from the total cell population. It was subjected to the process of normalization, dimension reduction, and clustering once again. With a resolution of 1, 4 clusters were identified (Supplementary Fig. 3D). According to the expressed markers of cells, clusters 0, 1, and 3 in stromal cells were regarded as cancer-associated fibroblasts (CAFs), while cluster 2 was considered endothelial cells. Based on the expressed markers of CAFs, clusters 0, 1, and 3 were regarded as inflammatory CAFs (iCAFs, markers: CXCL14, MT2A), myofibroblast (myCAFs, markers: RGS5, ACTA2, MYL9), and antigen-presenting CAFs (apCAFs, markers: CD74, HLA-DRA, and HLA-DPA1), respectively (Fig. 4D). The differential genes in each CAFs cluster were showed using heatmap plot (Supplementary Fig. 3E). Then, 15 cluster was considered as myeloid cells (markers: CD14, CD68), while 17 and 19 clusters were regarded as T cells (markers: CD3D, CD3E). The final clusters and annotations can be found in Fig. 4E. The differential gene expression of each cluster cell group was shown in Supplementary Table 1. CellChat analysis showed diverse and distinct interactions among these cell types (Fig. 4F). There were different ligand-receptor modes between C2 and CAFs, as well as between immune cells (Fig. 4G). The secretion of APP, CD99, COL1A1, COL6A1, COL6A3, FN1, MDK, in CAFs, was primarily directed toward CD74, CD99, SDC1, SDC4 receptors on the surface of C2. SPP1 was mainly produced by myeloid cells, which acted on the (ITGAV + ITGB1) receptor on the surface of C2. The MDK secreted by T cells was predominantly directed at the SDC4 and (ITGA6 + ITGB1) receptors on the surface of C2. CellChat analysis of signaling pathways demonstrated that MK, THBS, ANGPTL, VISFATIN, JAM, and ncWNT signaling pathways from tumor microenvironment were involved in regulating the characteristics of C2 (Fig. 4H, Supplementary Fig. 3F). These results demonstrated that modulating the characteristics of CSCs was complex and involved the alternation of multiple molecules and signaling pathways, and the role of CAFs and immune cells in the process should be given more attention.

Examining the stem cell landscape. (A) The activity of TFs in C2 and other clusters in BLCA. (B) The activation of main pathways in C2 and other clusters in BLCA. (C) The changes in metabolism of C2 and other clusters in BLCA. (D) Stromal cells are subdivided into iCAF, myCAF, apCAF, and endothelial cells (upper). The major markers expressed in iCAF, myCAF, and apCAF (low). (E) The final clusters and annotations for BLCA. (F) CellChat analysis displays communications among these cell types. The impact is greater with a thicker line. (G) CellChat is used to determine selected ligand-receptor interactions between immune cells, CAFs, and C2. The size of each circle is what indicated P-values. The color gradient is a representation of the level of interaction; blue and red colors are the smallest and largest values, respectively. (H) The heatmap exhibits the incoming and outgoing signaling patterns in C2, CAFs, and immune cells. The deeper color indicates more weight and strength of the regulation of the signaling pathway
Integrative construction of a consensus CSCLPI based on machine learning
The differential genes in C2 were abstracted (Supplementary Table 2) and the expression matrices of genes with adjusted P < 0.05 in the TCGA cohort were obtained.
We have performed functional enrichment analysis for the 341 genes to investigate their biological signatures. As shown in the Supplementary Fig. 2B, the results demonstrated that they were mainly involved in the regulation of cell cycle et al. We conducted a univariate Cox analysis and uncovered 29 potential CSCs-related genes that have a significant correlation with the prognosis of BLCA (Supplementary Table 3). The integration of machine learning was used to study the 29 potential CSCs-related prognostic genes. 99 prediction models were fitted in all cohorts by using the leave-one-out cross-validation framework. The results of calculating the C-index of each model across all cohorts were presented in Fig. 5A. Accordingly, the prognostic model developed by the RSF method was found to be the optimal model with the highest average C-index of 0.659 (Fig. 5A). The importance of variables was also evaluated (Fig. 5B), and the variables that contributed the most to the model were selected, including LSM3, S100A10, SPINT1, SPTSSB, and VPS29. A multivariate Cox analysis was conducted on five genes and it was determined that S100A10 (HR:1.90, 95%CI: 1.12 to 3.21, P = 0.016) and SPINT1 (HR: 2.25, 95%CI: 1.36 to 3.70, P = 0.001) were significantly associated with the prognosis of BLCA (Fig. 5C). The CSC-like prognostic index (CSCLPI) was constructed as follows: CSCLPI = (-0.498 * expression of LSM3) + (0.645 * expression of S100A10) + (0.811 * expression of SPINT1) + (-0.269 * expression of SPTSSB) + (-0.472 * expression of VPS29). TCGA samples were classified into the high CSCLPI and low CSCLPI groups using the median value of 1.045 as the cutoff value of the model. The KM survival analysis revealed a difference in survival between the CSCLPI subgroups (Fig. 5D). In the TCGA-BLCA training cohort, the area under the ROC curve based on the CSCLPI for predicting the 1-, 2- and 3-year survival was 0.749, 0.696 and 0.65, respectively (Fig. 5E). In the external validation cohorts, CSCLPI was also developed based on the five genes, and 1.045 was considered as the cutoff value to classify samples. GSE13507 and GSE48075 cohorts had distinct survival differences based on KM survival analysis (Fig. 5F), but GSE32894 and GSE48276 cohorts did not (Fig. 5F). In GSE13507, the area under the ROC curve for predicting the 1-, 2- and 3-year survival was 0.75, 0.627 and 0.656, respectively. In GSE32894, it was 0.765, 0.755 and 0.706, respectively.

Construction and validation of CSCLPI. (A) The C-index of a total of 99 prediction models from all datasets is based on 10 integrative machine learning algorithms using the LOOCV framework. The RSF method was utilized to construct the model, which had the highest average C-index of 0.659. (B) Ranking of variables’ importance. (C) The results of multivariate Cox regression analysis, and independent prognostic factors were determined. (D) The result of the KM survival analysis of CSCLPI in the TCGA cohort. (E) The results of the ROC curve for CSCLPI in predicting 1-year, 2-year, and 3-year survival in the TCGA cohort. (F) The results of the KM survival analysis of CSCLPI in the validation cohorts. (G) The results of the ROC curve for CSCLPI in predicting 1-year, 2-year, and 3-year survival in the validation cohorts
In GSE48075, it was 0.652, 0.733 and 0.815, respectively. In GSE48276, it was 0.615, 0.552 and 0.513, respectively (Fig. 5). The 1-year AUC value of CSCLPI was compared against those of a stem cell signature [36] and an EMT-related gene signature [37] in BLCA. In Supplementary Fig. 4, it is shown that the 1-year AUC value of a stem cell signature and an EMT-related gene signature were 0.71 and 0.659, respectively, whereas the CSCLPI was 0.749. These findings demonstrated the good performance of the CSCLPI in predicting the prognosis of patients with BLCA.
Differences in mRNAsi, gene mutation, small molecular drug sensitivity, and functional enrichment between CSCLPI subgroups
The OCLR algorithm was employed to calculate mRNAsi of each BLCA patient by analyzing their gene expression profiles. It was discovered that mRNAsi was significantly higher in the low CSCLPI group (P < 0.001) (Fig. 6A), and mRNAsi was negatively linked with the CSCLPI score (Spearman correlation = − 0.32, P < 0.001) (Fig. 6B). According to KM analysis, patients with high-mRNAsi in BLCA had a longer OS than those with low-mRNAsi (Fig. 6C).

Certain characteristics analysis for CSCLPI subgroups. (A) The comparison of the mRNAsi between CSCLPI subgroups. ***: p < 0.001. (B) The Spearman correlation of the CSCLPI score with mRNAsi. (C) The KM survival analysis showed that BLCA patients with higher mRNAsi had a better prognosis. (D) The waterfall plot demonstrated mutational genes in the high CSCLPI group (upper) and in the low CSCLPI group (low). (E) Summary for mutation in the high CSCLPI group (upper) and in the low CSCLPI group (low). (F) Small molecular drug sensitivity analysis between CSCLPI subgroups. (G) GO analysis of differential genes between CSCLPI subgroups for the top ten enrichment. (H) KEGG analysis for the top ten enrichment
The results from gene mutation analysis demonstrated that the gene mutation frequency in the high CSCLPI group was higher than that of the low CSCLPI group (95.89% vs. 94.59%). The top 10 mutational genes in the high CSCLPI group contain TTN, TP53, ARID1A, MUC16, KMT2D, KDM6A, RYR2, SYNE1, EP300, and KMT2C (Fig. 6D), which also have a mutation rate of over 20%. The top 10 mutational genes in the low CSCLPI group comprise of TP53, TTN, KMT2D, MUC16, KDM6A, SYNE1, ARID1A, PIK3CA, FLG, and KMT2C (Fig. 6D), which also have a mutation rate of over 20%. The most common mutation type in both groups was missense-mutation, followed by nonsense mutation and splice site (Fig. 6E). The correlation between mutated genes was analyzed in CSCLPI subgroups (Supplementary Fig. 5A). It was found that in the high CSCLPI group, RTK − RAS, NOTCH, Hippo and TP53 signaling pathways were highly and abnormally activated, while in the low CSCLPI group, RTK − RAS, NOTCH, Hippo, TP53, WNT and PI3K signaling pathways were highly and abnormally activated (Supplementary Fig. 5B).
We picked 10 drugs with the most significant P-value based on the results of small molecular drug sensitivity analysis. The findings showed that high CSCLPI group was more sensitive to belinostat, blebbistatin, brivanib, and daporinad, while low CSCLPI group was more sensitive to canertinib, dasatinib, gefitinib, ibrutinib, lapatinib, and myriocin (Fig. 6F).
Functional enrichment was carried out on the differential genes (low-CSCLPI vs. high-CSCLPI) between CSCLPI subgroups. The results of the GO analysis showed that the up-regulated genes were involved in regulation of extracellular matrix (Fig. 6G). KEGG analysis demonstrated that the up-regulated genes were available to regulate ECM − receptor interaction, and the PI3K − Akt signaling pathway et al. (Fig. 6H). These results suggested that there may be a difference in molecular regulation between CSCLPI subgroups.
Impact of tumor microenvironment on biological signatures between CSCLPI subgroups
At first, the differences in tumor-infiltrating associated cells between CSCLPI subgroups were assessed using ESTIMATE scores. The findings indicated that the high-CSCLPI group had a higher stromal score than the low-CSCLPI category, but there were no differences in immune score between the subgroups (Fig. 7A). We analyzed the types of immune cells infiltrating through ssGSEA. It’s surprising that there was no significant difference in the infiltration of other immune cells between the CSCLPI subgroups, aside from macrophages and regulatory T cells in the high CSCLPI group were higher (Fig. 7B). The immune-related pathways in the high CSCLPI group included cell chemokine receptor (CCR), MHC class I, para-inflammation, and Type I IFN Response, while in the low CSCLPI group, Type II IFN Response was found to be involved in immune regulation (Fig. 7C). TIDE can be used to predict the response to immunotherapy, and T cell dysfunction score can be used to evaluate the function of T cells. The T cell dysfunction score in the high CSCLPI group was found to be higher (Fig. 7D). However, there was no distinction in TIDE score between CSCLPI subgroups (Supplementary Fig. 6A). These findings indicated that immunosuppression was present in the high CSCLPI group, which was one of the causes of poor prognosis in patients with BLCA. There may be no significant difference in the efficacy of immunotherapy between CSCLPI subgroups. Furthermore, we found out that the high CSCLPI group had a higher CAF score than the low CSCLPI group (Fig. 7D). The EPIC analysis further showed that the high CSCLPI group had a more significant CAF score (Fig. 7E). Thus, stromal cells (mainly CAFs) may regulate the differences in biological signatures between CSCLPI subgroups.

Tumor microenvironment analysis. (A) ESTIMATE analysis between CSCLPI subgroups. The differences in stromal (left) and immune scores (right). (B-C) The immune cell infiltration and immune-related pathways made distinctions between CSCLPI subgroups. (D) TIDE analysis between CSCLPI subgroups. The differences in the dysfunction score (left) and CAFs score (right). (E) The differences in CAFs score between CSCLPI subgroups were calculated using EPIC analysis. (F) CSCLPI subgroups have distinct expression of cancer-associated fibroblast-specific markers. (G) A consensus clustering analysis was conducted on BLCA samples using markers specific to cancer-associated fibroblasts. The optimal groups were determined to be two. (H) The tNES analysis was based on F1 and F2. (I) The difference in CSCLPI score between F1 and F2. (J) F2 had a higher number of patients in the high CSCLPI group
NS: not significantly. *: P < 0.05; **: P < 0.01; ***: P < 0.001; ****: P < 0.0001
The expression of cancer-associated fibroblast-specific markers in BLCA was analyzed. These markers were found to be significantly expressed in the high CSCLPI group (Fig. 7F). Using consensus clustering analysis, TCGA samples were grouped according to cancer-associated fibroblast-specific markers. The best number of groups was two (Fig. 7G, Supplementary Fig. 6B), referred to as fibroblast group 1 (F1) and fibroblast group 2 (F2). The tNES analysis indicated that grouping had an excellent performance in F1 and F2 (Fig. 7H). By comparing the CSCLPI between F1 and F2, we found that F2 had a higher CSCLPI score (Fig. 7I), and the patients in the high CSCLPI group were more present in F2 (Fig. 7J). The findings indicated that the CAFs were responsible for some of the differences in biological signatures between CSCLPI subgroups in BLCA.
Construction and testing of a nomogram
Clinical characteristics were crucial in determining prognosis. The heatmap showed the relationship between five key genes and clinical characteristics (Fig. 8A). CSCLPI, age, gender, tumor stage, T stage, and N stage were evaluated in univariate and multivariate Cox regression analyses in order to uncover independent risk factors that influence prognosis (Fig. 8B and C). The multivariate Cox regression analysis showed that CSCLPI (HR:3.4305, 95%CI: 1.5896 − 7.4037, P = 0.0017) and N stage (HR: 2.3804, 95%CI: 1.1237 − 5.0429, P = 0.0236) were important risk factors for the prognosis of patients with BLCA. CSCLPI, age, gender, tumor stage, T stage, and N stage were the foundations of the nomogram construction, which was convenient to comprehensively evaluate the 1-year, 2-year, 3-year and 5-year prognosis of patients with BLCA (Fig. 8D). The calibration curves and DCA curves for the nomogram of 1-year, 2-year, 3-year and 5-year prognostic prediction were depicted (Fig. 8E and F). Furthermore, the C-index of the nomogram was 0.743 (95% CI: 0.665–0.821). According to the results, the nomogram was able to predict accurately.

Construction of a predictive nomogram. (A) The heatmap showed the relationship between five key genes and clinical characteristics. (B-C) Univariate and multivariate Cox regression analyses results of age, gender, tumor stage, T stage, N stage, and CSCLPI. (D) A nomogram for predicting the prognosis of 1 year, 2, 3, and 5 years for patients with BLCA. The prediction of the second patient in the TCGA cohort was depicted. (E-F) The calibration curves and DCA curves of 1-, 2-, 3- and 5-year prognostic prediction for the nomogram. For calibration curves, the more the blue solid line and the black dotted line coincide, the more accurate the prediction was. For DCA curves, if the curve exceeded the horizontal and vertical axes, it implied that the model had certain clinical predictive utility
Discussion
CSCs can be found in almost all solid tumors [38], and they were involved in regulating multiple cancer hallmarks, which were closely related to tumorigenesis, heterogeneity, progression, recurrence, treatment resistance etc. Both the strong plasticity of CSCs and the complex interactions between CSCs and the tumor microenvironment were responsible for the results [39]. CSCs in BLCA have been identified for several years. However, the characteristics of CSCs and their crosstalk with other cells in the microenvironment are still indistinct. In this study, we identified CSCs from malignant epithelial cells in BLCA using the CytoTRACE method based on single-cell sequencing data. The activation of transcription factors and related signaling pathways, as well as changes in metabolism within CSCs, were explored. The clusters of CAFs and immune cells were identified to reveal the crosstalk between CSCs and CAFs, as well as immune cells. Then, we screened the hub genes related to CSCs using multiple machine learning methods, and constructed and validated a CSCs prognostic model called CSCLPI. Its biological signatures were investigated and was found to have a good performance in predicting prognosis. Thus, CSCLPI is a reliable indicator for assessing progression, treatment, and patients’ prognosis of BLCA.
The abnormal activation of TFs was an essential step in tumor initiation and progression. In the study, E2F1, E2F2, E2F3, E2F4, and TFAM were significantly activated in CSCs in BLCA. They were discovered to play a role in regulating the properties of CSCs in numerous cancers. In rectal cancer stem cells, DLGAP1-AS2 can enhance radiation resistance by communicating with E2F1, which upregulates CD151 expression by activating the AKT/mTOR/cyclinD1 pathway [40]. In laryngeal squamous cell carcinoma, LINC00319 promotes the properties of cancer stem cells by upregulating HMGB3 through E2F1 [41]. In BLCA, lee et al. have discovered that E2F1 is strongly linked to the progression from NMIBC to MIBC, and the transcriptional events triggered by E2F1-EZH2-SUZ12 may be responsible for cancer aggressiveness and chemotherapy resistance [42]. The activation of E2F1 in cells treated with anticancer drugs resulted in the expression of CSCs-related genes, indicating the important role in modulating cancer stem properties. E2F2 participating in the regulation of cancer stem properties was found to be controlled by miRNA [43, 44], but with limited mechanisms. Yang et al. have revealed that in colorectal cancer, the E2F3-MEX3A-KLF4 axis is crucial in coordinating cancer stem cell self-renewal and differentiation [45]. E2F3 has methylation sites and is required for liver CSCs self-renewal and activation of Wnt/β-catenin [46]. rtcisE2F acts as a scaffold for IGF2BP2 and E2F3 mRNA to stabilize the latter, which promotes liver CSCs self-renewal and initiates liver tumorigenesis and metastasis by activating Wnt/β-catenin. CD133- osteosarcoma cells gain cancer-stem-cell-like characteristics by being suppressed through the activation of E2F4 signaling [47]. The mechanism behind E2F4 signaling regulation is still unclear. Wen et al. have reported that blocking mitochondrial function by silencing TFAM markedly reduced tumor-initiating potential of colon cancer cells [48]. The expression of cancer stem cells associated with genes downstream of Wnt/β-catenin signaling becomes decreased by blocking TFAM. These previous studies demonstrated that some members of the E2F family were involved in the maintenance of CSCs and were associated with cancer-related biological signatures. It is important to focus on their role in regulating CSCs characteristics in BLCA.
In addition, many intracellular signaling pathways and extracellular factors have been shown to be very important regulators of CSCs. In BLCA, intracellular signaling pathways that have been proven to be implicated in modulating the development and progression of bladder CSCs include related to epithelial-mesenchymal transition (EMT) (such as Wnt/β-catenin, Notch, and transforming growth factor-beta (TGF-β)), the sonic hedgehog signaling pathway, the PI3K/AKT pathway, MAPKs, and the JAK-STAT pathway [49]. These pathways could serve as promising therapeutic options for stopping BC progression. Our study revealed that the activity of PI3K/AKT and MAPK pathways was abnormally high in regulating CSCs of BLCA. PRDX2 has been shown to be upregulated significantly in bladder tumors and cisplatin-resistant bladder tumor cell lines, and it plays a role in regulating tumor stemness and EMT through the PI3K-AKT pathway [50]. Furthermore, overexpression of HER2 triggers the release of IL-8, which promotes stem cell-like phenotypes by activating the MAPK signaling pathway in BLCA [51]. The mechanism behind the regulation of PI3K/AKT and MAPK pathways needs further investigation in BLCA. Tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), T cells, CAFs, and other immune cells are the main extracellular factors that regulate CSCs [52]. CAFs stimulate stemness by activating the WNT and NOTCH pathways. Canonical WNT is the primary pathway that governs CSCs and promotes stemness, but NOTCH signaling has also been implicated in stem cell maintenance in several cancers [52]. Our study also found that the WNT signaling pathway was highly activated in CSCs, which was secreted by iCAF. In addition, the ANGPTL and THBS pathways from myCAF and apCAF, as well as the MK pathways from iCAF and myCAF, were determined to be involved in modulating CSCs in BLCA. Ma et al. have found that interferon secreted from bladder cancer cells can induce SLC14A1+ CAFs, which in turn promote stemness of BLCA via WNT5a/β-catenin pathway [53]. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment were responsible for managing the effects of myeloid cells on CSCs. Currently, it has been discovered that STAT3 and NF-κB signaling pathways are very prominent in MDSCs in promoting cancer cell stemness [38]. The identities of the MDSC-derived factors in many cancers are still emerging. In the study, we found GALECTIN signaling pathway derived from myeloid cells was implicated in inducing CSCs in BLCA. However, the mechanism is still indistinct. Different T cell subsets are responsible for maintaining CSCs [38]. The main pathways comprise PI3K/AKT, EMT, TGF-β, NF-κB and p38 MAPK pathways. In the study, we found JAM signaling pathway derived from T cells was involved in modulating CSCs in BLCA. However, the mechanism is still unclear. Our study also determined the interacted molecules on the cell surface between CSCs, CAFs, and immune cells, providing a deeper insight into the investigation of regulation on CSCs by factors from the tumor microenvironment.
A prognostic model (named CSCLPI) was constructed based on machine learning, and five key genes (LSM3, S100A10, SPINT1, SPTSSB, and VPS29) were screened using the RSF method. Our findings were consistent with previous studies that patients with higher mRNAsi indices had better prognosis [54, 55], despite the common understanding that CSCs are responsible for tumorigenesis, metastasis, and recurrence. We speculate that the discrepancy is the result of multiple factors. Firstly, the significant heterogeneity in markers and models that describe cancers among CSCs is a factor. Secondly, the sustainment and activation of CSCs is not only dependent on the tumor itself, but also on the microenvironment. Immune cells and CAFs are important factors. In Supplementary Fig. 5, we found the Notch, Wnt and Hippo signaling pathways were highly and abnormally activated in the low-CSCLPI group, which were associated with activation and development of CSCs (that may be associated with higher mRNAsi). In addition, we found that the differences in the infiltration of immune cells (such as regulatory T cells, macrophage) and in the CAFs. They were involved in the regulation of biological signature of CSCLPI. These results also demonstrated the complexity of the tumor development and the importance of tumor microenvironment regulation. It has been established that an increase in S100A10 expression is associated with poorer outcomes and/or chemoresistance in multiple cancer types, including lung, breast, ovary, pancreas, gallbladder, colorectum, and leukemia [56]. Especially in breast, it can induce the expression levels of stem cell-related genes [57]. SPINT1 expression was discovered to be upregulated in breast cancer and had a significant correlation with a poorer prognosis [58]. S100A10 and SPINT1 expression were identified as being linked to the regulation of CSCs in BLCA and the prognosis of patients, which can be promising therapy targets for CSCs. The CSCLPI constructed and validated in the study was found to be linked to prognosis and has a good performance in predicting prognosis in BLCA. Using multiple methods, it was proven that there were no differences in the infiltration of immune cells between the CSCLPI subgroups, which resulted in no difference in their response to immunotherapy. But our research showed that the CSCLPI subgroups had distinct infiltration of CAFs, which contributed to the difference in biological signature and drug treatment. Thus, CSCLPI can be used in clinic to predict the prognosis and chemotherapy response of patients with BLCA.
It is inevitable that this study will include several limitations. First, the limited clustering of immune cells prevented us from revealing the interactions between CSCs and other immune cells (e.g. Neutrophils, NK cells). Second, the data in our research were acquired from public databases rather than our own database. It would be better to verify CSCLPI using a clinical dataset from our own center. Third, the biological functions of S100A10 and SPINT1 related to CSCs characteristics in BLCA should be explored in vivo and in vitro in the future.
Conclusion
Taken together, in the study, cancer stem cells from malignant epithelial cells, cancer-associated fibroblasts, and immune cells in BLCA were identified, and the characterization of the stem cell landscape was revealed. A consensus CSCLPI for BLCA with 10 machine-learning algorithms was developed and validated, and its prognostic value and biological signature were revealed. We suggest that our stemness model has prospective clinical implications for prognosis prediction and drug treatment evaluation.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- BLCA:
-
Bladder Cancer
- NMIBC:
-
Non-Muscle Invasive
- MIBC:
-
Muscle Invasive
- CSCs:
-
Cancer Stem Cells
- ECs:
-
Epithelial Cells
- RSF:
-
Random Survival Forest
- CSCLPI:
-
Cancer Stem Cell-Like Prognostic Index
- CAFs:
-
Cancer-Associated Fibroblasts
- mRNAsi:
-
Messenger RNA expression-based stemness index
- TF:
-
Transcription Factor
References
Richters A, Aben KKH, Kiemeney LALM. The global burden of urinary bladder cancer: an update. World J Urol. 2020;38(8):1895–904.
Lobo N, Shariat SF, Guo CC, Fernandez MI, Kassouf W, Choudhury A, et al. What is the significance of variant histology in Urothelial Carcinoma? Eur Urol Focus. 2020;6(4):653–63.
Lopez-Beltran A, Cookson MS, Guercio BJ, Cheng L. Advances in diagnosis and treatment of bladder cancer. BMJ. 2024;384:e076743.
Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A. 2009;106(33):14016–21.
Shen H, Blijlevens M, Yang N, Frangou C, Wilson KE, Xu B, et al. Sox4 expression confers bladder Cancer Stem Cell properties and predicts for poor patient outcome. Int J Biol Sci. 2015;11(12):1363–75.
Zhu D, Wan X, Huang H, Chen X, Liang W, Zhao F, et al. Knockdown of Bmi1 inhibits the stemness properties and tumorigenicity of human bladder cancer stem cell-like side population cells. Oncol Rep. 2014;31(2):727–36.
Wang H, Mei Y, Luo C, Huang Q, Wang Z, Lu G-M, et al. Single-cell analyses reveal mechanisms of Cancer Stem Cell maintenance and epithelial-mesenchymal transition in recurrent bladder Cancer. Clin Cancer Res. 2021;27(22):6265–78.
Ma L, Chen H, Yang W, Ji Z. Crosstalk between mesenchymal stem cells and Cancer stem cells reveals a Novel stemness-related signature to predict prognosis and immunotherapy responses for bladder Cancer patients. Int J Mol Sci. 2023;24(5).
Lai H, Cheng X, Liu Q, Luo W, Liu M, Zhang M, et al. Single-cell RNA sequencing reveals the epithelial cell heterogeneity and invasive subpopulation in human bladder cancer. Int J Cancer. 2021;149(12):2099–115.
Kim W-J, Kim E-J, Kim S-K, Kim Y-J, Ha Y-S, Jeong P, et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol Cancer. 2010;9:3.
Sjödahl G, Lauss M, Lövgren K, Chebil G, Gudjonsson S, Veerla S, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. 2012;18(12):3377–86.
Guo CC, Bondaruk J, Yao H, Wang Z, Zhang L, Lee S, et al. Assessment of Luminal and basal phenotypes in bladder Cancer. Sci Rep. 2020;10(1):9743.
Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2):152–65.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM et al. Comprehensive integration of single-cell data. Cell. 2019;177(7).
Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol. 2021;39(5):599–608.
Lai Y, Ye J, Chen J, Zhang L, Wasi L, He Z, et al., et al. UPK3A: a promising novel urinary marker for the detection of bladder cancer. Urology. 2010;76(2):e5146–11.
Zhu L, Liu X, Zhang W, Hu H, Wang Q, Xu K. MTHFD2 is a potential oncogene for its strong association with poor prognosis and high level of immune infiltrates in urothelial carcinomas of bladder. BMC Cancer. 2022;22(1):556.
Unoki M, Kelly JD, Neal DE, Ponder BAJ, Nakamura Y, Hamamoto R. UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. Br J Cancer. 2009;101(1).
Ascione CM, Napolitano F, Esposito D, Servetto A, Belli S, Santaniello A, et al. Role of FGFR3 in bladder cancer: treatment landscape and future challenges. Cancer Treat Rev. 2023;115:102530.
Sanguedolce F, Zanelli M, Palicelli A, Bisagni A, Zizzo M, Ascani S et al. HER2 expression in bladder Cancer: a focused view on its Diagnostic, Prognostic, and predictive role. Int J Mol Sci. 2023;24(4).
Font A, Taron M, Gago JL, Costa C, Sánchez JJ, Carrato C, et al. BRCA1 mRNA expression and outcome to neoadjuvant cisplatin-based chemotherapy in bladder cancer. Ann Oncol. 2011;22(1):139–44.
García-Perdomo HA, Usubillaga-Velasquez JP, Zapata-Copete JA, Reis LO. Mutations in CDKN2A and the FGFR3 genes on bladder cancer diagnosis: a systematic review and meta-analysis. World J Urol. 2019;37(10):2001–7.
Rasteiro AM, Sá E, Lemos E, Oliveira PA. Gil Da Costa RM. Molecular markers in urinary bladder Cancer: applications for diagnosis, prognosis and therapy. Vet Sci. 2022;9(3).
Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–82.
Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367(6476):405–11.
Badia-I-Mompel P, Vélez Santiago J, Braunger J, Geiss C, Dimitrov D, Müller-Dott S, et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinform Adv. 2022;2(1):vbac016.
Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D, et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at single-cell level. Cancer Discov. 2022;12(1):134–53.
Liu Z, Liu L, Weng S, Guo C, Dang Q, Xu H, et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat Commun. 2022;13(1):816.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–8.
Racle J, Gfeller D. EPIC: a Tool to Estimate the proportions of different cell types from bulk gene expression data. Methods Mol Biol. 2020;2120:233–48.
Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN et al. Machine learning identifies stemness features Associated with Oncogenic Dedifferentiation. Cell. 2018;173(2).
Liu B, Zhan Y, Chen X, Hu X, Wu B, Pan S. Weighted gene co-expression network analysis can sort cancer-associated fibroblast-specific markers promoting bladder cancer progression. J Cell Physiol. 2021;236(2):1321–31.
Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021;22(6).
Chen X, Yin Y, He Y, Meng F, Zhao J, Liu F, et al. The prognostic significance and clinical relevance of stem cell characteristic in bladder cancer. Heliyon. 2024;10(3):e24858.
Cao R, Yuan L, Ma B, Wang G, Qiu W, Tian Y. An EMT-related gene signature for the prognosis of human bladder cancer. J Cell Mol Med. 2020;24(1):605–17.
Chen P, Hsu W-H, Han J, Xia Y, DePinho RA. Cancer Stemness meets immunity: from mechanism to Therapy. Cell Rep. 2021;34(1):108597.
Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25(1):20.
Xiao S-Y, Yan Z-G, Zhu X-D, Qiu J, Lu Y-C, Zeng F-R. LncRNA DLGAP1-AS2 promotes the radioresistance of rectal cancer stem cells by upregulating CD151 expression via E2F1. Transl Oncol. 2022;18:101304.
Yuan L, Tian X, Zhang Y, Huang X, Li Q, Li W, et al. LINC00319 promotes cancer stem cell-like properties in laryngeal squamous cell carcinoma via E2F1-mediated upregulation of HMGB3. Exp Mol Med. 2021;53(8):1218–28.
Lee S-R, Roh Y-G, Kim S-K, Lee J-S, Seol S-Y, Lee H-H, et al. Activation of EZH2 and SUZ12 regulated by E2F1 predicts the Disease Progression and aggressive characteristics of bladder Cancer. Clin Cancer Res. 2015;21(23):5391–403.
Feliciano A, Garcia-Mayea Y, Jubierre L, Mir C, Hummel M, Castellvi J, et al. miR-99a reveals two novel oncogenic proteins E2F2 and EMR2 and represses stemness in lung cancer. Cell Death Dis. 2017;8(10):e3141.
Lin Q-Y, Wang J-Q, Wu L-L, Zheng W-E, Chen P-R. miR-638 represses the stem cell characteristics of breast cancer cells by targeting E2F2. Breast Cancer. 2020;27(1):147–58.
Yang X, Li G, Tian Y, Wang X, Xu J, Liu R, et al. Identifying the E2F3-MEX3A-KLF4 signaling axis that sustains cancer cells in undifferentiated and proliferative state. Theranostics. 2022;12(16):6865–82.
Chen Z, Huang L, Wang K, Zhang L, Zhong X, Yan Z, et al. rtcisE2F promotes the self-renewal and metastasis of liver tumor-initiating cells via N6-methyladenosine-dependent E2F3/E2F6 mRNA stability. Sci China Life Sci. 2022;65(9):1840–54.
Feng J, Lan R, Cai G, Lin J. TREX1 suppression imparts cancer-stem-cell-like characteristics to CD133- osteosarcoma cells through the activation of E2F4 signaling. Int J Clin Exp Pathol. 2019;12(4):1134–53.
Wen Y-A, Xiong X, Scott T, Li AT, Wang C, Weiss HL, et al. The mitochondrial retrograde signaling regulates wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 2019;26(10):1955–69.
Abugomaa A, Elbadawy M, Yamawaki H, Usui T, Sasaki K. Emerging roles of Cancer Stem cells in bladder Cancer progression, Tumorigenesis, and resistance to Chemotherapy: a potential therapeutic target for bladder Cancer. Cells. 2020;9(1).
Li P, Wang W, Zhu B, Wang Y, Li J, Wang C, et al. PRDX2 regulates stemness contributing to cisplatin resistance and metastasis in bladder cancer. Environ Toxicol. 2024;39(5):2869–80.
Zhou Q, Jin P, Liu J, Li S, Liu W, Xi S. HER2 overexpression triggers the IL-8 to promote arsenic-induced EMT and stem cell-like phenotypes in human bladder epithelial cells. Ecotoxicol Environ Saf. 2021;208:111693.
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–38.
Ma Z, Li X, Mao Y, Wei C, Huang Z, Li G et al. Interferon-dependent SLC14A1 + cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell. 2022;40(12).
Zheng H, Liu H, Li H, Dou W, Wang J, Zhang J, et al. Characterization of stem cell landscape and identification of stemness-relevant prognostic gene signature to aid immunotherapy in colorectal cancer. Stem Cell Res Ther. 2022;13(1):244.
Wang W, Xu C, Ren Y, Wang S, Liao C, Fu X, et al. A Novel Cancer Stemness-Related signature for Predicting Prognosis in patients with Colon adenocarcinoma. Stem Cells Int. 2021;2021:7036059.
Saiki Y, Horii A. Multiple functions of S100A10, an important cancer promoter. Pathol Int. 2019;69(11):629–36.
Yanagi H, Watanabe T, Nishimura T, Hayashi T, Kono S, Tsuchida H, et al. Upregulation of S100A10 in metastasized breast cancer stem cells. Cancer Sci. 2020;111(12):4359–70.
Wu Q, Yin G, Luo J, Zhang Y, Ai T, Tian J, et al. Comprehensive Analysis of the expression and Prognostic Value of SPINT1/2 in breast carcinoma. Front Endocrinol (Lausanne). 2021;12:665666.
Acknowledgements
We greatly appreciate the TCGA program, GEO database, ESTIMATE database, and Tumor Immune Dysfunction and Exclusion for providing the open-source data.
Funding
The study was supported by Natural Science Foundation of Lianshui (No. 202314), Natural Basic Research Plan of Huai’an (No. HABL2023103), the National Natural Science Foundation of China (No. 82204866), the Natural Science Foundation of Jiangsu Province (No. BK20231163), and the Pengcheng Talent - Medical Young Reserve Talent Training Project (No. XWRCHT20220012).
Author information
Authors and Affiliations
Contributions
WQW, GTL, JML, HW, and LL designed the study. GTL, JML, HW, LCW, FFZ, LQW, LX, YD, LL and WQW collected the data. GTL, JML, HW, LCW, FFZ, LQW, LX, YD, LL and WQW analyzed the data. GTL, JML, and HW wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
The study protocols were approved by the Ethical Committee Review Board of the 900th Hospital of Joint Logistic Support Force (Fuzhou, Fujian, China).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, G., Lin, J., Wang, H. et al. Characterization of the stem cell landscape and identification of a stemness-associated prognostic signature in bladder cancer. Cancer Cell Int 24, 299 (2024). https://doi.org/10.1186/s12935-024-03465-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-024-03465-4