
Abstract
Background
Familial hemophagocytic lymphohistiocytosis (FHL) is a genetically heterogeneous autosomal recessive hyper-inflammatory syndrome which needs early accurate diagnosis and appropriate treatment to prevent complications and early mortality. Recently, it was reported that mutations in STXBP2 gene are linked to FHL type 5 (FHL-5).
Case Presentation
We report a Sri Lankan neonate who presented with low Apgar scores at birth, abdominal distension, and hepatosplenomegaly, followed by lethargy, poor sucking and rapid decompensation with wide spread activation of inflammation within 48 h of birth. Her elder sibling also had a similar presentation during early neonatal period and deceased at two weeks of age with no diagnosis. Unfortunately, the index case deceased at 14 days of age following multi-organ dysfunction and severe metabolic acidosis. Targeted gene panel followed by reflex exome sequencing revealed a novel likely pathogenic homozygous variant in the STXBP2 gene (NM_001272034.1:c.1141-2A > G) which confirmed the diagnosis of autosomal recessive FHL-5.
Conclusion
Early diagnosis of FHL type 5 using genetic analysis and timely treatment are difficult in the absence of family history due to a wide spectrum of clinical manifestations. However both early diagnosis and treatment doesn’t alter the long term prognosis. So genetic counselling would be the better option.
Similar content being viewed by others


Background
Familial hemophagocytic lymphohistiocytosis (FHL) is a rare and potentially fatal autosomal recessive hyper-inflammatory syndrome [1]. FHL is caused by genetic mutations resulting in defective cell cytotoxicity. There are five disease related genes: mutation in chromosome 9q (FHL1/ type 1), PFR1 (FHL2/ type 2), UNC13D (FHL3/ type3), STX11 (FHL4/type 4) and STXBP2 (FHL5. type 5) [2]. But four genes in which pathogenic variants are causative have been identified: FHL types 2, 3 and 4 are associated with defective function of perforin, Munc13-4 and syntaxin-11 respectively, whilst type 5 (STXBP2) is associated with a defect in Munc18-2 [3, 4]. The defective genes in FHL lead to ineffective T lymphocyte and natural killer cell degranulation by inhibition of cytotoxic granule components impairing their fusion machinery [5]. Ultimately, the disease is characterized by inefficient removal of target cells and uncontrolled activation of macrophages and T cells [1]. The incidence of FHL is about 0.12 to 0.15 per 100,000 children per year [1, 5, 6]. FHL-5 secondary to causative variants of STXBP2 accounts for 10% of all FHL cases [1, 5, 6]. FHL type 5 due to mutation by causative variants of STXBP2 with neonatal presentation is extremely rare [8] and most children present between 2 and 6 months of age [9]. Due to atypical and wide range of presentation of FHL, most neonates who do not have a positive family history are diagnosed either late in the course of the disease or following death [10]. FHL is clinically characterized by recurrent fever, hepatosplenomegaly, cytopenias, hypertriglyceridemia, hypofibrinogenemia, hyperferritinemia and hemophagocytosis in bone marrow and the clinical features mimic severe sepsis [11]. In the newborn with hydrops foetalis, the disease resembles congenital infections. The mortality of untreated patients can be as high as 95%. The recognized treatment modalities include immune-chemotherapy for disease control and hematopoietic stem cell transplantation. The authors report a neonate with foetal onset of FHL5 disease with hydrops foetalis and a novel homozygous likely pathogenic variant in the STXBP2 gene.
Case presentation
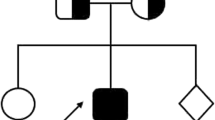
The index case is a female newborn found to have low Apgar scores and abdominal distention at birth. She was the product of third pregnancy of non-consanguineous parents. The first pregnancy ended as a spontaneous-onset first trimester miscarriage. Her elder child, born following second pregnancy, died during the neonatal period with no diagnosis. The elder sibling had a similar presentation during the early neonatal period and later developed severe sepsis and metabolic acidosis (Fig. 1).

Pedigree from the FHL families with STXBP2 gene mutation
The index child was born by elective caesarian section weighing 3 kg at term. Mother did not report of any medical disorders or infections complications during the pregnancy. After low Apgar scores improved following initial resuscitation, she continued to have mild tachypnea and abdominal distension. She had poor sucking, fever, neonatal jaundice and marked hepatosplenomegaly on day 1.
The baby was treated as for severe sepsis with intravenous antibiotics despite negative septic screen. However, given the previous unexplained neonatal death, this child was thoroughly investigated for a differential diagnosis including metabolic disorders. Over the course, the baby deteriorated with worsening of jaundice and needed ventilatory care.
Her liver functions were abnormal with evidence of widespread activation of inflammation: INR (International normalized ratio)—6.23, ALT-586 u/L, AST-5865 u/L, serum bilirubin 362 micromol/L, GGT-248u/L, serum albumin-2.2 g/dl, APTT – 48 s. Renal functions were also deranged: blood urea—146 mg/dL, serum creatinine—2.2 mg/dL). Haematological parameters showed progressive increase in white blood cells (17 × 103 mm/L with neutrophils -30%), thrombocytopenia (platelets—8 × 103 mm/L), and anemia (haemoglobin—6.3 g/dL). C-reactive protein (CRP) was 116 mg/dL. Blood smear showed features of infection with toxic granules and pancytopenia. Metabolic investigations showed: lactic acidosis and increased urinary excretion lactic acid and 4-hydroxyphenyllactic acid. Serum alpha-fetoprotein was very high (14900 ng/ml). TORCH screening was negative. Screening for Hepatitis B and hepatitis C was negative. Ferritin and triglyceride were not performed. Blood and CSF cultures remained negative while urine culture grew Klebsiella. Biochemical studies of cerebrospinal fluids were normal. Ultrasound chest and abdomen showed pleural effusion and ascites respectively and the findings were in keeping with hydrops fetalis. Brain ultrasound was normal.
Despite intensive management with ventilatory care, inotropes, blood and its derivatives, and potent intravenous antibiotics, the patient deteriorated leading to multi-organ dysfunction and severe metabolic acidosis and deceased on day 14 while receiving ventilator care.
Given the clinical picture of the patient, genetic analysis of an extended genetic panel was requested at CENTOGENE AG. Genomic DNA was enzymatically fragmented and regions of interest were enriched using DNA capture probes targeted against the coding regions along with flanking ± 10 intronic bases and known pathogenic/likely pathogenic variants of the panel genes. The libraries were subsequently sequenced on an Illumina platform.
Initial evaluation of a gene panel was negative, thus, the analysis was extended to other genes from the exome. A homozygous splicing variant in the STXBP2 [Chr19(GRCh37)] gene NM_001272034.1:c.1141-2A > G was detected. The variant is extremely rare and absent in control patients. In silico tools predict that this variant disrupts the highly conserved acceptor splice site of exon 14. This variant is classified as likely pathogenic based on the American College of Medical Genetics and Genomic guidelines (ACMG). The following criteria for variant classification were applied: PVS1 (null variant) and PM2 (absent or very rare in controls, i.e. gnomAD) [12]. The annotation of the variant and the predictions tools in silico used are present in Table 1. For splicing mutations, Ada- and RF-scores, together with conservation were considered. At the time of the initial report, we were also using SSF, MaxEnt, HSF prediction tools that all supported the likely splice effect as reported (threshold differences are > 5% for SSF, > 15% for MaxEnt or > 10% for HSF) (Table 2).
Parents were offered genetic studies to confirm heterozygosity of the variant and arrangements were made to follow up with genetic counseling and prenatal diagnosis.
Both babies presented with similar clinical features in the early neonatal period, but index baby exhibited clinical features at birth that the first deceased baby discharged on day 2 following neonatal assessment with normal ultrasound abdomen, only showed after readmission on the following day due to poor sucking and mild jaundice. Table 1 described the clinical and genetic information of both deceased babies.
Discussion and conclusions
Familial hemophagocytic lymphohistiocytosis is characterized by uncontrolled inflammation. Fetal onset FHL is considered to be the most severe form and carries high mortality [10]; however due to extreme rarity, disease characteristics of fetal onset neonatal FHL is not well described [13].
FHL is caused by genetic mutations resulting in defective cell cytotoxicity and it is divided in five different subtypes, depending on the affected gene [2]. Our newborn had a novel homozygous likely pathogenic variant in the STXBP2 gene (type 5) which is associated with defect in Munc18-2 [3, 4]. STXBP2 is a part of the Sec/ Munc proteins that are important for the assembly and disassembly of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex and the control of membrane fusion. The exact role of STXBP2 in the cytotoxic granule pathway is not presently described in depth, however, the previous studies detailed that the protein is also involved in the regulation of intracellular granule trafficking in the epithelial cells, neutrophils, and mast cells [14]. Deficiency of this protein leads to defective cytotoxic activity and regulation of epithelial cells and neutrophils, and removal of target cell and uncontrolled activation of macrophages and T lymphocytes which causes immune dysfunction [1].The uncontrolled inflammation in FHL is associated with very high interleukin 6, interleukin 8 and tumor necrosis factors 8 levels with markedly reduced natural killer cell function in the neonate [15].
Although clinical features in FHL usually present during the first year of life, the affected individuals might not reveal symptoms until later in childhood or even into adulthood [16]. Well-recognised clinical features of FHL include prolong fever, enlarged liver and/or spleen, skin rash, lymph node enlargement, breathing problems, kidney abnormalities, heart problems, and cytopenias. FHL is also associated with an increased risk for certain haematological malignancies such as leukemia and lymphoma [17, 18].
FHL5 typically presents within the first months or years of life and occasionally, in utero and is associated with atypical features such as severe diarrhoea, abnormal bleeding, and early onset neurological abnormalities including hearing loss [14, 19]. The index case exhibited symptoms at birth which include, poor sucking, less active, tachypneoa, hepatosplenomegaly, hydrops foetalis, cytopenia and jaundice. Subsequently prolong fever, septicaemia and bleeding tendency secondary to liver dysfunction and thrombocytopenia were observed. Although she did not have renal and cardiac abnormalities at birth, she developed metabolic acidosis and multi-organ failure with the progression of the disease.
The diagnosis of hemophagocytic lymphohistiocytosis (HL) can be established If 1 and/or 2 of below criteria are fulfilled: (1) a genetic test identifying a mutation in one of the genes known to be involved in FHL, (2) at least five out of the following eight clinical and laboratory features: fever, splenomegaly, pancytopaenia, hypertriglyceridemia, hypofibrinogenaemia, hyperferritinaemia, haemophagocytosis on bone marrow smear, spleen or lymph node biopsy, decreased or absent natural killer cell activity, and elevated blood levels of CD25 [19, 20]. The genetic study confirmed the diagnosis in our newborn.
Treatment depends on a number of factors, including the severity of symptoms, the age of onset, and the underlying cause of haemophagocytosis. Allogeneic hematopoietic cell transplantation is thought to be curative for FHL. Better outcomes have been observed following early marrow transplantation in confirmed or suspected patients with FHL [21]. Before the era of allogeneic hematopoietic cell transplantation, patients were usually treated with chemotherapy and/or immunotherapy such as corticosteroids, cyclosporine, etoposide, or anti-thymocyte globulin to destroy excess immune cells, and to counteract life-threatening hyper-inflammatory syndrome. Since the authors did not suspect the possibility of FHL at birth and the diagnosis was only made retrospectively based on genetic studies, these treatment options could not be offered to the reported patient.
All forms of FHL, whether treated early or not, carry a high mortality rate. The long-term prognosis of familial forms without treatment is poor, with a median survival of less than 2 to 6 months after appearance of symptoms. Even with proper treatment, estimated five-year survival would only be 21–26% in patients with FHL [22].
We offered genetic counselling with the help of multidisciplinary team which includes Consultant Genetics, Neonatologist, Obstetrician and Gynecologist, Psychiatrist and Chemical pathologist on regular time basis. They accepted the loss and now have a normal social life. Furthermore, we explained the various options to have a baby including adopting a baby. Although we offered the genetic studies on parents and explained the future intrauterine diagnosis, parents would like to wait 2 years to plan next pregnancy and are planning genetic studies, but their economic condition and religious background are barriers that prevent them to conduct further genetic studies at the moment. Till then, parents agreed to follow-up counselling with our team.
Early diagnosis of FHL type 5 is often difficult in the absence of family history and also due to a wide range of clinical presentations. Early diagnosis is crucial in timely treatment. Exome sequencing has the potential to genetically diagnose these patients with heterogeneous clinical presentations.
Availability of data and materials
The data that support the findings of this case report are available from Medical Records Department, Batticaloa Teaching Hospital, but restrictions apply to the availability of these data, which were used under license for the current report and so are not publicly available. Data are, however, available from the authors upon reasonable request and with permission of Medical Records Department, Batticaloa Teaching Hospital, Sri Lanka.
Abbreviations
- FHL:
-
Familial Hemophagocytosis Lymphohistiocytosis
- HL:
-
Hemophagocytosis Lymphohistiocytosis
- CSF:
-
Cerebrospinal fluid
- B/C:
-
Blood culture
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- GGT:
-
Gamma-glutamyl transferase
- APTT:
-
Activated Partial Thromboplastin Time
- TORCH screen:
-
Toxoplasmosis, other, rubella cytomegalovirus, herpes simplex, and HIV screen
References
Tang, X.MM, Guo, XM, Li, QMD, et al Familial hemophagocytic lymphohistiocytosis type 5 in a Chinese Tibetan patient caused by a novel compound heterozygous mutation in STXBP2, October 2019 - Volume 98 - Issue 43 - p e17674.
Allen CE, McClain KL. fl-ILH: becoming a blended family. Blood. 2014;124:1210–1.
Bryceson YT, Rudd E, Zheng C, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110(6):1906–15. https://doi.org/10.1182/blood-2007-02-074468.
Valentina Cetica, Alessandra Santoro, Kimberly C Gilmour, STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5, J Med Gene.t2010sep:47(9):595–600.
Zhao XW, Gazendam RP, Drewniak A, et al. Defects in neutrophil granule mobilization and bactericidal activity in familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) syndrome caused by STXBP2/Munc18-2 mutations. Blood. 2013;122:10911. https://doi.org/10.1182/blood-2013-03-494039.
Meeths M, Horne A, Sabel M, et al. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer. 2015;62:34652.
Aricò M, Janka G, Fischer A. et al.Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. Leukemia. 1996;10(2):197–203.
Bechara E, Dijoud F, de Saint BG, Bertrand Y, Pondarré C. Hemophagocytic lymphohistiocytosis with Munc13-4 mutation: a cause of recurrent fatal hydrops fetalis. Pediatrics. 2011;128(1):e251–4.
Suzuki N Morimoto A Ohga S Kudo K Ishida Y Ishii E; HLH/LCH Committee of the Japanese Society of Pediatric Hematology. Characteristics of hemophagocytic lymphohistiocytosis in neonates: a nationwide survey in Japan J Pediatr 20091552235–80..
Whaley BF. Familial hemophagocytic lymphohistiocytosis in the neonate. Adv Neonatal Care. 2011;11(2):101–7. https://doi.org/10.1097/ANC.0b013e318210d02c.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Isaacs H Jr. Fetal and neonatal histiocytoses. Pediatr Blood Cancer. 2006;47(2):123–9.
Pagel J, Beutel K, Lehmberg K, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocyticlymphohistiocytosis type 5 (FHL5). Blood. 2012;119(25):6016–24. https://doi.org/10.1182/blood-2011-12-398958.
Iwatani S, Uemura K, Mizobuchi M, et al. Familial Hemophagocytic Lymphohistiocytosis Presenting as Hydrops Fetalis. AJP Rep. 2015;5(1):e22–4. https://doi.org/10.1055/s-0034-1544110.
Baothman A, Almalki H, Abumelha K, Alshegifi A, Baashar A. Type 5 Familial Hemophagocytic Lymphohistiocytosis in a Seven-year-old Girl Post Second Bone Marrow Transplantation with Failure to Thrive: STXBP2 Novel Mutation. Cureus. 2019;11(11):e6246. Published 2019 Nov 27. doi:https://doi.org/10.7759/cureus.6246
Kenneth L McClain, MD, PhD. Clinical features and diagnosis of hemophagocytic lymphohistiocytosis. UpToDate. July 29, 2015.
Familial hemophagocytic lymphohistiocytosis. Genetics Home Reference. November 2014; http://ghr.nlm.nih.gov/condition/familial-hemophagocytic-lymphohistiocytosis.
Kejian Z, Alexandra HF, Judith J, et al. Hemophagocytic Lymphohistiocytosis, Familial. Gene Reviews. January 17, 2013; http://www.ncbi.nlm.nih.gov/books/NBK1444/.
Zhang L, Zhou J, Sokol L. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control. October 2014;21(4):301–12. https://doi.org/10.1177/107327481402100406.
Michniacki TF, Mulcahy Levy JM, Quinones RR, Giller RH. Successful correction of familial hemophagocyticlymphohistiocytosis using prenatal genetic testing and preemptive hematopoietic stem cell transplantation. Bone Marrow Transplant. 2018;53(2):223–4. https://doi.org/10.1038/bmt.2017.219.
Schwartz RA. Lymphohistiocytosis (Hemophagocytic Lymphohistiocytosis). Medscape. 2016; http://emedicine.medscape.com/article/986458-overview.
Acknowledgements
The authors would like to thank our multidisciplinary team which includes Consultant Psychiatrists, Genetics and Chemical pathologist in participating in the counseling session for the parents in addition to us.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
VT, MT, MTRJ and MLMJ led clinical management of the babies and parents. VT performed literature survey, wrote manuscript and edited the manuscript. KD performed literature survey and edited the manuscript. EJ performed laboratory evaluation and edited manuscript. CP and VS performed genetic testing and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from both of the patient’s parents.
Consent for publication
Written informed consent for publication of the patient’s identifying images or other personal or clinical details was obtained from both of the patient’s parents. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Thadchanamoorthy, V., Jayatunga, M.T.R., Dayasiri, K. et al. Exome sequencing detected an extremely rare case of foetal onset familial haemophagocytic lymphohistiocytosis type 5 presenting with hydrops foetalis. BMC Med Genomics 14, 50 (2021). https://doi.org/10.1186/s12920-021-00897-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00897-z