
Abstract
Background
Physical symptoms and aversion induced by opioid withdrawal strongly affect the management of opioid addiction. YTH N6-methyladenosine (m6A) RNA binding protein 1 (YTHDF1), an m6A-binding protein, from the periaqueductal gray (PAG) reportedly contributes to morphine tolerance and hyperalgesia. However, the role of YTHDF1 in morphine withdrawal remains unclear.
Methods
A naloxone-precipitated morphine withdrawal model was established in C57/BL6 mice or transgenic mice. YTHDF1 was knocked down via adeno-associated virus transfection. Combined with the results of the single-cell RNA sequencing analysis, the changes in morphine withdrawal somatic signs and conditioned place aversion (CPA) scores were compared when YTHDF1 originating from different neurons in the ventrolateral periaqueductal gray (vlPAG) was knocked down. We further explored the role of inflammatory factors and transcription factors related to inflammatory response in morphine withdrawal.
Results
Our results revealed that YTHDF1 expression was upregulated in the vlPAG of mice with morphine withdrawal and that the knockdown of vlPAG YTHDF1 attenuated morphine withdrawal-related somatic signs and aversion. The levels of NF-κB and p-NF-κB were reduced after the inhibition of YTHDF1 in the vlPAG. YTHDF1 from vlPAG inhibitory neurons, rather than excitatory neurons, facilitated morphine withdrawal responses. The inhibition of YTHDF1 in vlPAG somatostatin (Sst)-expressing neurons relieved somatic signs of morphine withdrawal and aversion, whereas the knockdown of YTHDF1 in cholecystokinin (Cck)-expressing or parvalbumin (PV)-expressing neurons did not change morphine withdrawal-induced responses. The activity of c-fos + neurons, the intensity of the calcium signal, the density of dendritic spines, and the frequency of mIPSCs in the vlPAG, which were increased in mice with morphine withdrawal, were decreased with the inhibition of YTHDF1 from vlPAG inhibitory neurons or Sst-expressing neurons. Knockdown of NF-κB in Sst-expressing neurons also alleviated morphine withdrawal-induced responses.
Conclusions
YTHDF1 originating from Sst-expressing neurons in the vlPAG is crucial for the modulation of morphine withdrawal responses, and the underlying mechanism might be related to the regulation of the expression and phosphorylation of NF-κB.
Similar content being viewed by others


Background
Opioid drugs are among the most powerful analgesics but also among the most addictive drugs. Patients treated with opioids or misusing them are vulnerable to developing physical dependence, which manifests with the emergence of withdrawal symptoms when the use of opioids is abruptly discontinued [1]. Dependence can lead to opioid seeking and contribute to addiction by perpetuating repeated exposures to avoid withdrawal symptoms. Chronic exposure to opioids results in conditioning, and opioid withdrawal can lead to aversion [2]. Alleviating or preventing withdrawal-induced somatic responses and aversion can be a strategy for developing treatments for opioid addiction.
The periaqueductal gray (PAG) is a major component of the pain modulatory system [3], and many reports have indicated that the PAG plays an important role in the processes of reward and conditioning, as well as in morphine withdrawal syndrome [4,5,6]. Previous studies have shown that the manipulation of the ventrolateral periaqueductal gray (vlPAG) could modulate the responses induced by morphine withdrawal [7,8,9]. N6-methyladenosine (m6A), the most prevalent internal RNA modification of mammalian messenger RNAs (mRNAs), is abundant in the nervous system and participates in the modulation of various neural functions [10,11,12]. YTHDF1, an m6A-binding protein, has been reported to mediate the process of learning and memory formation facilitated by m6A [13]. Our previous study revealed that chronic morphine exposure could increase m6A methylation in the vlPAG and that the knockdown of YTHDF1 in the vlPAG alleviated morphine analgesic tolerance and morphine-induced hyperalgesia (MIH) [14]. We reasonably speculate that the somatic symptoms associated with morphine withdrawal and withdrawal-induced aversion can be mediated by the altered expression level of YTHDF1 in the vlPAG.
The PAG contains different types of neurons that utilize glutamate, γ-aminobutyric acid (GABA), opioids, and other neurotransmitters [15]. Different neuronal populations in the PAG may exert different effects on the same physiological process [9, 16]. Samineni et al. showed that selective chemogenetic activation of glutamatergic neurons or inhibition of GABAergic neurons in the vlPAG suppresses nociception, whereas inhibition of glutamatergic neurons or activation of GABAergic neurons in the vlPAG facilitates nociception [16]. Our recent study revealed that the inhibition of vlPAG GABAergic neurons, rather than the activation of GABAergic neurons or the inhibition or activation of glutamatergic neurons, could significantly attenuate morphine withdrawal-induced conditioned place aversion (CPA) [9]. The above studies suggest that GABAergic neurons in the vlPAG are likely related to the modulation of morphine withdrawal responses. Similarly, different subtypes of functional neurons are not necessary to develop the same or similar reactions under certain circumstances or stimuli. Among the different subtypes of neurons in the PAG, tachykinin 1-expressing neurons, but not Sst-expressing neurons, were found to be required for itch-induced scratching behavior [17]. Thus, whether YTHDF1 from different subtypes of neurons in the vlPAG might exert different effects on the modulation of morphine withdrawal responses is a question to be answered.
Neuroinflammation in the vlPAG has been revealed to be involved in the development of morphine tolerance [18, 19]. Proinflammatory cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), and the transcription factor nuclear factor-κB (NF-κB) related to inflammatory response contribute to the development of morphine tolerance [20,21,22,23]. Studies have revealed that the severity of morphine withdrawal responses is related to the neuroinflammatory state [24,25,26]. Doyle et al. reported that increased glial cell reactivity and neuroinflammation contribute functionally to morphine withdrawal in male mice [24]. Manipulation of different pathways in the central nervous system could alter the level of neuroinflammation and modulate morphine withdrawal symptoms and conditioned place preference (CPP) [25, 26]. Our previous study demonstrated that the YTHDF1‒TRAF6 pathway regulates the neuroinflammatory response and contributes to morphine tolerance and hyperalgesia in the PAG [14]. However, the exact effects of YTHDF1 on proinflammatory cytokines and their interactions within different neuron subtypes in the vlPAG during morphine withdrawal remain unclear.
Therefore, we investigated the level of YTHDF1 in the vlPAG of mice with naloxone-precipitated morphine withdrawal. We knocked down YTHDF1 expression in the vlPAG of wild-type and transgenic mice subjected to morphine withdrawal to explore the effect of YTHDF1 on somatic signs associated with morphine withdrawal and morphine withdrawal-induced aversion. We hypothesized that the modulation of morphine withdrawal symptoms and aversion could be achieved by regulating the levels of YTHDF1 and mediating the inflammatory responses in inhibitory neurons originating from the vlPAG.
Materials and methods
Animals
Male C57/BL6 mice (8–10 weeks) were purchased from the Institute of Experimental Animals of Guangdong Medicine Experimental Animal Center. YTHDF1flox/flox (YTHDF1fl/fl) mice were constructed by GemPharmatech (Nanjing, China) and were obtained from the laboratories of Prof. Ruihua Xu and Prof. Huaiqiang Ju as gifts. The genotype of the YTHDF1fl/fl mice was verified in Xu and Ju’s research [27] and in our previous study [14]. Gad2-Cre mice (JAX: 028867) were purchased from the Jackson Laboratory. All the mice were housed under a standard 12-h light/dark cycle (light from 7:00 a.m. to 7:00 p.m.) at constant room temperature (25 ± 1 °C) with food and water available ad libitum. All experimental procedures were approved by the Use Committee of Sun Yat-sen University and the Animal Care Committee (No. L102012020000X) and were conducted in accordance with the guidelines of the National Institutes of Health (NIH).
Naloxone-precipitated morphine withdrawal model
The mice were injected (s.c.) with increasing doses of morphine twice per day (at 8 am and 4 pm) for four consecutive days. The escalating strategy for morphine treatment was as follows: 10 mg/kg on the 1st day, 20 mg/kg on the 2nd day, 30 mg/kg on the 3rd day, and 40 mg/kg on the 4th day. The mice were returned to their home cages after each morphine injection. On day 5, the mice were injected with 40 mg/kg morphine (s.c.), and 1 h later, 1 mg/kg naloxone (s.c.) was administered to induce morphine withdrawal.
Evaluation of morphine withdrawal somatic signs
The somatic signs of morphine withdrawal were evaluated as described below (Fig. 1a). On days 1 and 2, each mouse was placed in a transparent box and allowed to freely explore it. The mice were randomly divided into two groups: the morphine withdrawal group and the control group. From day 3 to day 7, the naloxone-precipitated morphine withdrawal model was established as described in the section on the naloxone-precipitated morphine withdrawal model. The mice in the control group were administered the same dose of morphine from day 3 to day 7, but the same volume of saline (s.c.) was used as a control to replace naloxone. Immediately after naloxone administration, the mice were individually placed in a transparent box, and withdrawal signs were evaluated over the course of 30 min. Jumps, paw tremors, teeth chattering (vacuous chewing), wet-dog shake, diarrhea, and loss of body weight were recorded as somatic signs of withdrawal. The body weights of the mice were recorded before the injection of naloxone and 60 min after the injection, and the percentage of body weight loss after morphine withdrawal was used as one evaluation measurement. The number of jumps was recorded quantitatively. Teeth chattering, paw tremors, wet-dog shake, and diarrhea were evaluated over 30-s periods, with one point assigned for the presence of each sign during each period. A global withdrawal score was calculated for each mouse by assigning a weighting factor to the various physical signs of withdrawal [28,29,30,31]. The global withdrawal score was calculated as follows: (number of jumps × 0.1) + (number of paw tremors × 0.1) + (number of teeth chattering events × 0.1) + (number of wet-dog shakes) + (number of diarrhea events × 2) + (loss of body weight percent (%) × 5). Drug injections and behavioral counts were performed by an operator blinded to the groups.

Establishment of a naloxone-precipitated morphine withdrawal model. a Experimental timeline of the test for physical signs of morphine withdrawal and the morphine withdrawal CPA test. b Schematic of the three-compartment apparatus applied for the CPA test. c The morphine withdrawal score (***P < 0.001, t test, n = 10) of WT mice was significantly increased after morphine withdrawal. d The CPA aversion score of WT mice was significantly reduced after morphine withdrawal (***P < 0.001, t test, n = 10). e The mean velocity did not differ significantly before and after morphine withdrawal (prep-aired, N.S. = not significant; Mann‒Whitney U test, n = 10; post-paired, N.S. = not significant; Mann‒Whitney U test; n = 10). f IHC images showing that the number of c-fos+ neurons increased after morphine withdrawal (scale bar = 100 μm), and the corresponding statistical result is shown (***P < 0.001, t test, n = 6). g–h Heatmaps showing the Ca2+ signals of morphine withdrawal model mice compared with those of normal control mice and the corresponding mean GCaMp6s signal along with the statistical results (***P < 0.001, t test, n = 6). i Representative images of GFP-labeled dendrites and statistical data for the density of dendritic spines showing that the density of dendritic spines was significantly greater after morphine withdrawal (***P < 0.001, t test, n = 6). j–l Representative traces of mIPSCs recorded from vlPAG neurons and statistical data for the average frequency and amplitude of mIPSCs showing an increased mIPSC frequency after morphine withdrawal (***P < 0.001, t test, n = 6). The data are shown as the means ± SEMs
Morphine withdrawal conditioned place aversion (CPA) test
The CPA test was performed in the CPA apparatus with another group of mice (Fig. 1a). The CPA apparatus consisted of two conditioning compartments and one central connecting compartment (Fig. 1b). The left compartments had walls with black‒white horizontal stripes and smooth floors, whereas the right compartments had walls with black‒white vertical stripes and frosted floors to distinguish between the two conditioning compartments. The grouping method was the same as that in the section describing the evaluation of somatic signs. On days 1 and 2, all the mice had free access to the entire apparatus for 15 min to reduce the effects caused by the experimental environment. Animals with a strong initial preference for either compartment (one compartment > 720 s) were eliminated from the study. We randomly assigned any one conditioning compartment as a paired compartment for each mouse to balance the CPA assay and avoid possible effects between the two conditioning compartments. From day 3 to day 7, we established the morphine withdrawal model as described in the section on the naloxone-precipitated morphine withdrawal model. On day 7, the mice were confined to the paired compartments for 1 h immediately after naloxone was injected subcutaneously. On day 8, the mice were allowed to explore the entire apparatus for 15 min. The CPA score was defined as the time (post-paired) spent in the paired compartment minus the time (pre-paired) spent in the paired compartment. Analysis software (JLBehv-CPPM-4, Jiliang Technology Co., Ltd., Shanghai, China) was used to record and analyze the time and movement path in the two conditioning compartments.
Surgeries and stereotaxic injections
Under continuous isoflurane inhalation anesthesia, the mouse was placed in a stereotaxic frame (RWD Life Science Co., Ltd.) with the bregma and lambda horizontal, and the body temperature was maintained at 36 °C with a heating pad. The stereotaxic coordinates for viral vector injection were as follows: anteroposterior (AP), − 4.45 mm; mediolateral (ML), ± 0.55 mm; and dorsoventral (DV), − 2.7 mm. After the administration of additional local analgesia with a 1 ml subcutaneous injection of 1% lidocaine above the skull, the skull was fully exposed and perforated with a stereotaxic drill. The virus was infused bilaterally through a microinjector with a 33G needle. The virus mixture (150 nl) was infused for 10 min. After infusion, the needle was retained at the injection site for more than 15 min before being withdrawn slowly.
Knockdown of YTHDF1
The shRNA target of YTHDF1 used in the study was GGAAATGCCCAACCTACTTCT. CMV-Cre along with AAV-DIO-YTHDF1-shRNA were stereotaxically injected into the bilateral vlPAG to knock down the expression of YTHDF1 in wild-type (WT) mice. CMV-Cre-EGFP was injected into the vlPAG to knock down the expression of YTHDF1 in YTHDF1flox/flox (YTHDF1fl/fl) mice. CamkIIα-Cre along with AAV-DIO-YTHDF1-shRNA were used to specifically knock down YTHDF1 in the vlPAG excitatory neurons of WT mice, whereas CamkIIα-Cre-EGFP was used in YTHDF1fl/fl mice. We applied Vgat-Cre with AAV-DIO-YTHDF1-shRNA in WT mice, Vgat-Cre-EGFP in YTHDF1fl/fl mice, and AAV-DIO-YTHDF1-shRNA in Gad2-Cre mice to knock down YTHDF1 in vlPAG inhibitory neurons. Sst-Cre, PV-Cre, or Cck-Cre along with AAV-DIO-YTHDF1-shRNA were used to knock down YTHDF1 in Sst-expressing, PV-expressing, or Cck-expressing neurons. We also constructed Vgat-YTHDF1-shRNA and Sst-YTHDF1-shRNA to knock down YTHDF1 in inhibitory neurons and Sst-expressing neurons for dendritic spine analyses. The mice were housed in the cages for 3 weeks without any other treatment after the virus injection to ensure stable transgene expression from the AAV. The verification of the specificity of Sst-Cre as a promoter and the knockdown efficiencies of CMV-YTHDF1-shRNA, CamkIIα-YTHDF1-shRNA, Vgat-YTHDF1-shRNA, and Sst-YTHDF1-shRNA are shown in Additional file 1: Fig. S1.
Calcium signal recordings
All the mice were anesthetized with pentobarbital sodium. AAV-hSyn-GCaMp6s was used for the nonselective detection of calcium signaling in neurons, and AAV-EF1α-DIO-GCaMp6s with Vgat-Cre or Sst-Cre were utilized to detect calcium signaling in inhibitory neurons or Sst-expressing neurons. An optical fiber (200-μm core, 0.37 NA; Inper, Inc.) was placed in a ceramic ferrule and inserted into the vlPAG (Additional file 1: Fig. S1). All the mice were individually housed and allowed to recover for 1 week after surgery. The 470-nm laser power was set to 40 μW. GCaMP fluorescence was detected by a fiber photometry system (Thinkerbiotech, China), and MATLAB software (R2021b) was used to analyze the calcium signals. The baseline fluorescence was acquired prior to each signal recording (5 min). The signals were normalized to the baseline to calculate △F/F0 = (Fsignal − Fbaseline)/Fbaseline.
Immunohistochemical (IHC) analyses
The mice were perfused with 4% paraformaldehyde under anesthesia. The brain tissues were cut into 20 μm-thick sections after 30% DEPC-sucrose dehydration at 4 °C. The tissue samples used for the c-fos analysis were prepared 1 h after the establishment of naloxone-precipitated morphine withdrawal, and the tissue samples used for other analyses were prepared 24 h after the establishment of morphine withdrawal. Primary antibodies against YTHDF1 (Proteintech, 26,787–1-AP, 1:200), c-fos (Abcam, ab222699, 1:200), NF-κB (Cell Signaling Technology, 8242 T, 1:200), p-NF-κB (Affinity Biosciences, AF2006, 1:100), IL-1β (Abcam, ab9722, 1:200), IL-6 (Affinity Biosciences, DF6087, 1:200), TNF-α (Affinity Biosciences, AF7014, 1:200), NeuN (Merck, MAB377, 1:200), GFAP (Abcam, ab4648, 1:100), and Sst (Abcam, ab111912, 1:100) diluted in hybridization solution were then incubated with the vlPAG sections at 4 °C overnight. The sections were subsequently incubated with fluorescein secondary antibodies conjugated with Cy3 or Alexa Fluor 488 at room temperature (approximately 26 °C) for 1 h. Finally, the sections were stained with DAPI and imaged using a confocal microscope (Nikon) equipped with a digital camera.
Western blotting
The proteins extracted from the vlPAG were quantified using a BCA protein assay kit, separated on SDS‒PAGE gels, and transferred onto PVDF membranes. TBST with 3% skim milk was applied to block the PVDF membranes with gentle shaking at room temperature (approximately 26 °C) for 1 h to avoid nonspecific binding. Primary antibodies against YTHDF1 (Proteintech, 26,787–1-AP, 1:1000), NF-κB (Cell Signaling Technology, 8242 T, 1:1000), p-NF-κB (Affinity Biosciences, AF2006, 1:1000), IL-1β (Abcam, ab9722, 1:1000), IL-6 (Affinity Biosciences, DF6087, 1:1000), TNF-α (Affinity Biosciences, AF7014, 1:1000) and GAPDH (Fude Biological Technology, FD0063, 1:1000) were then incubated with the PVDF membranes at 4 °C overnight. After the membrane was rinsed three times with TBST, it was incubated for 1 h with an anti-rabbit IgG secondary antibody at room temperature. Immunostained bands were detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore, WBKLS0500) and an Image Quant LAS 4000 mini system (GE Healthcare). Band intensities were analyzed using ImageJ and normalized to those of GAPDH.
RNA extraction and real-time quantitative polymerase chain reaction (PCR)
TRIzol was used to extract total DNA from the vlPAG, and reverse transcription was performed using the polymerase chain reaction (PCR) production kit (Accurate Biology, AG 11706) according to the manufacturer’s instructions. The primer pair of YTHDF1 used for qRT‒PCR was as follows: forwards: 5′-ACCTGTCCAGCTATTACCCG-3′, reverse: 5′-TGGTGAGGTATGGAATCGGAG-3′. The reaction cycling conditions were as follows: initial denaturation at 95 °C for 3 min, followed by 40 thermal cycles of 10 s at 95 °C, 20 s at 58 °C, and 10 s at 72 °C. The ratio of mRNA expression in the vlPAG tissues was analyzed via the 2−ΔΔCT method.
Dendritic spine analysis
One month before the morphine withdrawal model was established, sparsely labeled viruses, Hysn-FLP:FDIO-EGFP, Vgat-FLP:FDIO-EGFP, and Sst-FLP:FDIO-EGFP, obtained from Brain Case Biotechnology Corporation (Shenzhen, China), were injected into the mouse vlPAG to label the morphology of the dendritic spines. We used a laser confocal microscope to capture images of dendritic spines in multilayer Z-axes, and reconstructed the dendritic spine images. The spines were classified into one of four morphological subtypes: filopodial, thin, stubby, or mushroom shaped. ImageJ was used (http://rsbweb.nih.gov/ij) to calculate the density of thin, stubby, and mushroom-shaped dendritic spines. Approximately 10 randomly selected neurons were analyzed per condition across two coverslips. The density of spines was scored in dendritic segments 10 μm in length. Finally, we counted and used the number of dendritic spines per 10 μm to describe the density of the dendritic spines.
Electrophysiological recordings
The mice were deeply anesthetized with pentobarbital sodium and decapitated. Coronal slices (250 μm thick) containing the vlPAG were prepared using a vibratome (VT1200S, Leica) in ice-cold cutting solution containing the following components: 110 mM choline chloride, 2.5 mM KCl, 1.3 mM NaH2PO4, 7 mM MgCl2·6H2O, 0.5 mM CaCl2·2H2O, 25 mM NaHCO3, and 20 mM D-glucose. Slices were recovered at 32 °C for 30 min in artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2·2H2O, 1.3 mM NaH2PO4, 25 mM NaHCO3, 1.3 mM MgCl2·6H2O, and 10 mM D-glucose, equilibrated with 95/5% O2/CO2. The brain slices were incubated at room temperature until they were transferred into a recording chamber.
AAV-EF1α-DIO-EGFP along with Vgat-Cre were used to label GABAergic neurons in the vlPAG. AAV-EF1α-DIO-EGFP along with Sst-Cre were used to label the Sst-expressing neurons in the vlPAG. A P97 micropipette puller was used to prepare borosilicate pipettes with a resistance of 3 to 5 megohms. A Multiclamp 700B amplifier and Digidata 1550A with pClamp 10.6 software (Molecular Devices) were used to perform electrophysiological recordings. Whole-cell voltage clamp recording was performed to measure mIPSCs. The mIPSCs were recorded using a bath solution that contained blockers of sodium channel currents (1 μM tetrodotoxin, Sigma, 554,412) and glutamatergic synaptic transmission (10 μM DNQX, Sigma, D0540), with the pipette solution containing 140 mM CsCl, 10 mM HEPES, 4 mM MgCl2·6H2O, 0.5 mM EGTA, 4 mM Na2ATP, 0.4 mM Na4GTP, and 10 mM QX-314 (pH 7.3, 297 mOsm). The recordings lasted for more than 10 min at a − 70-mV holding potential for the mIPSCs, ensuring that 1–2 min of stable data were recorded for analysis. Clampfit v10.6 software (Molecular Devices) was used to initially process the data from the voltage clamp recordings. mIPSCs were detected and analyzed using MiniAnalysis (Synaptosoft). The threshold for mIPSC detection was 10 pA. Automatic detection was manually verified post hoc. Statistical analysis and data plotting were performed using GraphPad Prism v9.2.
Bioinformatic analysis of single-cell RNA sequencing data
The raw single-cell RNA sequencing (scRNA-seq) data from GBM samples (GSE240626) [32, 33] were obtained from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. Codes are available on request. The R package “DoubletFinder” algorithm (https://github.com/chris-mcginnisucsf/DoubletFinder) [34] was applied to computationally detect doublets. Doublets were removed from each sample individually, with an expected doublet rate of 0.05 and default parameters used otherwise. The remaining cells that survived the doublet-detection filtering criteria were single cells. The R package Seurat (version 3.2.3, https://satijalab.org/seurat) [35] was subsequently used to combine and convert the expression information of the remaining cells to the Seurat object. Next, the cells that had fewer than 101 UMIs, expressed fewer than 501 genes, or had more than 15% UMIs linked to mitochondrial genes were removed. Next, the NormalizeData and ScaleData functions of the Seurat package were run. We integrated the scRNA-seq data with the Harmony algorithm (https://github.com/immunogenomics/harmony) [36] to correct for the batch effect. We obtained cell clusters using the FindCluster function of the Seurat package and visualized cells with the uniform manifold approximation and projection (UMAP) algorithm.
We annotated the cell clusters based on their average gene expression of canonical markers, including ependymocyte (Dnah11), ASTRO (Slca10), microglial (Cx3cr1), endothelial (Pecam), OLIGOS (S100b/Opalin), inhibitory neuron (Gad2), and excitatory neuron (slc17a6/Slc17a8/Tph2/Gabra6) markers, which were consistent with the original studies.
Statistical analysis
The data were analyzed using SPSS (SPSS version 25.0; IBM, USA). All the data are presented as the means ± standard errors of the means (SEMs). All the statistical analyses were two-tailed comparisons. Two-sample independent t tests and Mann‒Whitney U tests were used to analyze the data, when appropriate. A P value less than 0.05 was considered statistically significant (N.S. = not significant, P > 0.05; *P < 0.05, **P < 0.01, and ***P < 0.001).
Results
YTHDF1 was upregulated in the vlPAG of mice with morphine withdrawal, and YTHDF1 knockdown relieved morphine withdrawal responses
We first established a naloxone-precipitated morphine withdrawal model to explore the effects of vlPAG YTHDF1 on morphine withdrawal-induced somatic signs and conditioned place aversion (CPA) (Fig. 1a–b). After chronic exposure to morphine, naloxone elicited physical signs of morphine withdrawal (Fig. 1c) and induced CPA (Fig. 1d) without affecting the ability of the animals to move (Fig. 1e). In the naloxone-precipitated morphine withdrawal model mice, the number of c-fos+ neurons was significantly increased in the vlPAG (Fig. 1f), indicating increased neuronal activation in the vlPAG. Using AAV-hSyn-GCaMp6s, we found that the Ca2+ signal in the vlPAG was markedly increased in morphine withdrawal model mice (Fig. 1g–h), which also indicated increased neuronal activity. The density of dendritic spines, marked by Hysn-FLP:FDIO-EGFP, was significantly greater in the morphine-withdrawal mice than in the control mice (Fig. 1i), indicating increased synaptic activity. After the nonselective detection of mIPSCs in vlPAG neurons, the results revealed that the frequency of mIPSCs increased in morphine withdrawal model mice (Fig. 1j–k) without a significant change in the amplitude of mIPSCs (Fig. 1l), indicating increased inhibitory synaptic transmission during morphine withdrawal.
Next, we explored the role of vlPAG YTHDF1 in morphine withdrawal. The PCR results revealed that the relative expression of the YTHDF1 mRNA in the vlPAG was increased in the morphine withdrawal model mice (Fig. 2a). Both the WB and IHC results revealed a significant increase in YTHDF1 expression in the morphine withdrawal model mice (Fig. 2b–c). We knocked down the expression of vlPAG YTHDF1 in wild-type (WT) mice and YTHDF1flox/flox (YTHDF1fl/fl) mice to further confirm the role of vlPAG YTHDF1 in morphine withdrawal responses. We injected CMV-Cre along with AAV-DIO-YTHDF1-shRNA into the vlPAG of WT mice to knock down YTHDF1. Downregulation of vlPAG YTHDF1 effectively alleviated all somatic signs of morphine withdrawal, including jumps, paw tremors, teeth chattering, wet-dog shakes, diarrhea, and loss of body weight, and lowered the morphine withdrawal score (Fig. 2d). The aversion score improved after the knockdown of vlPAG YTHDF1 (Fig. 2e). The knockdown of YTHDF1 resulted in a decreased number of c-fos + neurons (Fig. 2f) and a reduced density of dendritic spines (Fig. 2g), indicating attenuated neuronal and synaptic activity in mice with YTHDF1 knockdown subjected to morphine withdrawal. Similar results were observed in the YTHDF1fl/fl mice. CMV-Cre-EGFP was used to conditionally knock down YTHDF1 in YTHDF1fl/fl mice. Knocking down vlPAG YTHDF1 in YTHDF1fl/fl mice also relieved the physical symptoms of morphine withdrawal and reduced the morphine withdrawal score (Fig. 2h), along with improving the aversion score (Fig. 2i). We also detected diminished c-fos signals (Fig. 2j) and a reduced dendritic spine density (Fig. 2k) in the YTHDF1fl/fl mice treated with CMV-Cre-EGFP. These data suggest that the downregulation of vlPAG YTHDF1 could be beneficial for the modulation of morphine withdrawal-related somatic signs and CPA and that this modulation might be relevant to neuronal and synaptic activity in the vlPAG.

vlPAG YTHDF1 was upregulated in morphine withdrawal model mice, and YTHDF1 knockdown attenuated morphine withdrawal responses. a The relative expression of the YTHDF1 mRNA was increased in morphine withdrawal model mice (***P < 0.001, t test, n = 6). b Western blotting revealed that YTHDF1 expression was increased in morphine withdrawal model mice (***P < 0.001, t test, n = 6). c IHC images showing that YTHDF1 expression was upregulated after morphine withdrawal. Scale bar = 100 μm. d The morphine withdrawal score (***P < 0.001, t test, n = 10) of WT mice was significantly lower after the knockdown of vlPAG YTHDF1. e Aversion was alleviated in WT mice after the knockdown of vlPAG YTHDF1 (***P < 0.001, t test, n = 10). f IHC images showing that the number of c-fos + neurons was decreased in WT mice after the knockdown of vlPAG YTHDF1 (scale bar = 100 μm), and the corresponding statistical results are shown (***P < 0.001, t test, n = 6). g The density of dendritic spines was significantly reduced in WT mice after the knockdown of vlPAG YTHDF1 (***P < 0.001, t test, n = 6). h The morphine withdrawal score (***P < 0.001, t test, n = 10) of the YTHDF1fl/fl mice was significantly lower after vlPAG YTHDF1 was knocked down. i Aversion was alleviated in YTHDF1fl/fl mice after the knockdown of vlPAG YTHDF1 (***P < 0.001, t test, n = 10). j IHC images showing that the number of c-fos + neurons was reduced in YTHDF1fl/fl mice after the knockdown of vlPAG YTHDF1 (scale bar = 100 μm), and the corresponding statistical results are shown (**P < 0.01, t test, n = 6). k The density of dendritic spines was significantly reduced in YTHDF1fl/fl mice after the knockdown of vlPAG YTHDF1 (***P < 0.001, t test, n = 6). The data are shown as the means ± SEMs
YTHDF1 from vlPAG inhibitory neurons facilitated morphine withdrawal responses
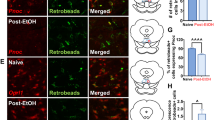
We reanalyzed the single-cell RNA sequencing (scRNA-seq) data from a previous study to examine the expression of the YTHDF1 gene in each cell type in the vlPAG [32, 33]. A total of 12,638 cell transcriptomes were retained for subsequent analysis and were annotated into 11 major cell types (Fig. 3a–c). We found that most neurons in the vlPAG that expressed YTHDF1 were either excitatory neurons or inhibitory neurons. We further verified this finding through the results of IHC shown in Fig. 3d–e, which indicated that YTHDF1 was co-expressed in CamkIIα + neurons or Vgat + neurons in the vlPAG. More information about the normalized expression of cell markers in each cell cluster is shown in Additional file 1: Fig. S2, and the expression of YTHDF1 in NeuN + , CX3CR1 + , and GFAP + neurons is shown in Additional file 1: Fig. S3.

Expression level of the YTHDF1 gene in different cell types in the vlPAG based on single-cell RNA-seq data. a UMAP plot of 12,638 single cells grouped into 11 major cell types in the vlPAG. Each dot represents a single cell, which is colored according to the cell type. b Feature plot of the normalized expression of the YTHDF1 gene in each cell type in the vlPAG; the depth of color from gray to blue represents low to high expression. c The expression level of the YTHDF1 gene in each cell cluster in the vlPAG. d–e IHC images depicting the co-expression of YTHDF1 in CamkIIα + (d) and Vgat + (e) neurons
As different subtypes of neurons in the vlPAG have been reported to possibly exert different effects [9], we next selectively knocked down YTHDF1 originating from inhibitory or excitatory neurons to investigate the roles of different vlPAG neurons in morphine withdrawal responses. First, we investigated the action of excitatory neurons in the vlPAG. AAV-CamkIIα-Cre along with AAV-DIO-YTHDF1-shRNA were utilized to specifically knock down YTHDF1 expression in excitatory neurons in WT mice. The results revealed that the somatic signs of morphine withdrawal were not alleviated (Fig. 4a), and the aversion scores did not differ significantly between the two groups (Fig. 4b). We also knocked down YTHDF1 originating from vlPAG excitatory neurons in YTHDF1fl/fl mice. Similarly, knocking down YTHDF1 in vlPAG excitatory neurons in YTHDF1fl/fl mice did not cause significant changes in the somatic signs of morphine withdrawal (Fig. 4c) or aversion scores (Fig. 4d).

The knockdown of YTHDF1 in CamkIIα + neurons in the vlPAG had no effect on the morphine withdrawal response, whereas the knockdown of YTHDF1 in Vgat + neurons in the vlPAG attenuated the morphine withdrawal response. a The morphine withdrawal score of WT mice was not significantly affected after the knockdown of YTHDF1 in CamkIIα + neurons (N.S. = not significant, t test, n = 10). b The aversion score of WT mice did not change significantly after the knockdown of YTHDF1 in CamkIIα + neurons (N.S. = not significant, t test, n = 10). c The morphine withdrawal score of YTHDF1fl/fl mice was not significantly affected after the knockdown of YTHDF1 in CamkIIα + neurons (N.S. = not significant, t test; n = 10). d The aversion score of YTHDF1fl/fl mice did not change significantly after the knockdown of YTHDF1 in CamkIIα + neurons (N.S. = not significant, t test, n = 10). e The morphine withdrawal score of WT mice was decreased (***P < 0.001, Mann‒Whitney U test, n = 10) after the knockdown of YTHDF1 in Vgat + neurons. f Aversion was alleviated in WT mice after the knockdown of YTHDF1 in Vgat + neurons (**P < 0.01, t test, n = 10). g The morphine withdrawal score of YTHDF1fl/fl mice was decreased (***P < 0.001, t test, n = 10) after the knockdown of YTHDF1 in Vgat + neurons. h Aversion was alleviated in YTHDF1fl/fl mice after the knockdown of YTHDF1 in Vgat + neurons (**P < 0.01, t test, n = 10). i The morphine withdrawal score of Gad2-Cre mice was decreased (***P < 0.001, t test, n = 10) after the knockdown of YTHDF1 in Vgat + neurons. j Aversion was alleviated in Gad2-Cre mice after the knockdown of YTHDF1 in Vgat + neurons (**P < 0.01, t test, n = 10). The data are shown as the means ± SEMs
We subsequently investigated the effect of YTHDF1 from inhibitory neurons on morphine withdrawal responses. AAV-Vgat-Cre along with AAV-DIO-YTHDF1-shRNA were injected into the vlPAG of WT mice to selectively knock down YTHDF1 expression in inhibitory neurons. The knockdown of YTHDF1 in vlPAG inhibitory neurons relieved the physical symptoms of morphine withdrawal, reduced the morphine withdrawal score (Fig. 4e), and improved the morphine withdrawal-induced CPA (Fig. 4f). We performed additional tests using YTHDF1fl/fl mice injected with AAV-Vgat-Cre and Gad2-Cre mice injected with AAV-DIO-YTHDF1-shRNA. The somatic signs of morphine withdrawal and CPA performance improved when the expression of YTHDF1 in inhibitory neurons was inhibited in both the YTHDF1fl/fl mice (Fig. 4g–h) and the Gad2-Cre mice (Fig. 4i–j). These results indicate that YTHDF1 originating from inhibitory neurons, but not excitatory neurons, in the vlPAG participates in the modulation of morphine withdrawal-related somatic signs and CPA.
YTHDF1 may modulate morphine withdrawal responses through NF-κB
Since YTHDF1 expressed in vlPAG inhibitory neurons regulates morphine withdrawal responses, the underlying mechanism is worthy of investigation. Our previous study demonstrated that the YTHDF1‒TRAF6 pathway contributes to morphine tolerance and hyperalgesia in the PAG by regulating proinflammatory cytokines and the transcription factor related to inflammatory response, including IL-1β, IL-6, TNF-α, and NF-κB [14]. Whether the interaction between YTHDF1 and proinflammatory cytokines or transcription factors related to inflammatory response from vlPAG inhibitory neurons affects the process of morphine withdrawal needs clarification. We analyzed the single-cell sequencing data to determine the expression levels of IL-1β, IL-6, TNF-α, and NF-κB in the vlPAG. As shown in Fig. 5a–i, only NF-κB, but not IL-1β, IL-6, or TNF-α, was found to be expressed in the inhibitory neurons. Thus, we next investigated the effect of YTHDF1-NF-κB from vlPAG inhibitory neurons on morphine withdrawal responses. In the morphine withdrawal model mice, Vgat-Cre and DIO-YTHDF1-shRNA were injected into the vlPAG to inhibit the expression of YTHDF1 in the inhibitory neurons. The levels of IL-1β, IL-6, TNF-α, and NF-κB were examined through IHC and western blotting after the inhibition of YTHDF1. After the inhibition of YTHDF1, the levels of NF-κB and p-NF-κB were reduced (Fig. 6a–c), whereas the expression of IL-1β, IL-6, or TNF-α was not significantly changed (Additional file 1: Fig. S4). The WB results revealed that the reduction in the p-NF-κB level was greater than that in the NF-κB level (Fig. 6c), implying that the phosphorylation level of NF-κB was also downregulated after the knockdown of YTHDF1. Thus, we speculate that YTHDF1 from vlPAG inhibitory neurons modulates morphine withdrawal responses by regulating the expression and phosphorylation of NF-κB.

Expression levels of the IL-1β, IL-6, TNF-α, and NF-κB genes in different cell types in the vlPAG based on single-cell RNA-seq data. a–d Violin plots showing the expression levels of the TNF-α (a), IL-1β (b), IL-6 (c), and NF-κB (d) genes in each cell cluster in the vlPAG. e–h Feature plots of the normalized expression of the TNF-α (e), IL-1β (f), IL-6 (g), or NF-κB (h) genes in each cell type in the vlPAG. (i) Dot plot showing the expression levels of YTHDF1, TNF-α, IL-1β, IL-6, and NF-κB in each cell cluster in the vlPAG

The levels of NF-κB and p-NF-κB were downregulated after the knockdown of YTHDF1 in Vgat + neurons in the vlPAG. a–b IHC images showing the co-expression of YTHDF1 and NF-κB (a) or p-NF-κB (b); the levels of NF-κB (a) and p-NF-κB (b) were reduced after the knockdown of YTHDF1 in Vgat + neurons in the vlPAG. c Western blotting revealed that the expression of NF-κB (***P < 0.001, t test, n = 6) and the ratio of p-NF-κB to NF-κB (*P < 0.05, t test, n = 6) were reduced after the knockdown of YTHDF1 in Vgat + neurons in the vlPAG. d IHC images showing the reduced expression of YTHDF1 in c-fos + neurons after the knockdown of YTHDF1 in Vgat + neurons in the vlPAG (scale bar = 100 μm), and the corresponding statistical results are shown (**P < 0.01, t test, n = 6). e–f Ca2+ signaling was reduced in morphine withdrawal model mice after the inhibition of YTHDF1 in vlPAG inhibitory neurons; the corresponding mean GCaMp6s signal along with the statistical results are shown (***P < 0.001, t test, n = 6). g The density of dendritic spines was significantly reduced after the knockdown of YTHDF1 in Vgat + neurons in the vlPAG (**P < 0.01, t test, n = 6). h–j Representative traces of mIPSCs recorded in vlPAG GABAergic neurons from morphine withdrawal model mice and statistical data for the average frequency and amplitude of mIPSCs show the decreased mIPSC frequency after the knockdown of YTHDF1 in vlPAG GABAergic neurons (**P < 0.01, t test, n = 6). The data are shown as the means ± SEMs
Next, we examined neuronal and synaptic activity after the inhibition of YTHDF1 from vlPAG inhibitory neurons. The co-expression of c-fos and YTHDF1 in inhibitory neurons was significantly reduced in morphine withdrawal model mice treated with Vgat-Cre along with DIO-YTHDF1-shRNA (Fig. 6d). We injected AAV-EF1α-DIO-GCaMp6s along with Vgat-Cre into the vlPAG to detect Ca2+ signals in inhibitory neurons, and the results revealed that after the downregulation of YTHDF1 from the vlPAG inhibitory neurons, the Ca2+ signal was obviously weakened (Fig. 6e–f), suggesting decreased neuroexcitability in the vlPAG neurons after the downregulation of YTHDF1 in inhibitory neurons. Through labeling with Vgat-FLP:FDIO-EGFP and knocking down YTHDF1 with Vgat-YTHDF1-shRNA, we found that the density of dendritic spines was reduced after the inhibition of YTHDF1 in inhibitory neurons (Fig. 6g). AAV-EF1α-DIO-EGFP along with Vgat-Cre were used to label GABAergic neurons in the vlPAG, followed by selective electrophysiological recordings. The results revealed that the frequency of mIPSCs in the vlPAG decreased when the expression of YTHDF1 in inhibitory neurons was knocked down, whereas the amplitude of mIPSCs remained the same (Fig. 6h–j), indicating the effect of YTHDF1 on the inhibitory synaptic transmission intensity that participates in the modulation of morphine withdrawal responses. These results demonstrated that YTHDF1 from vlPAG inhibitory neurons might alter the excitability of neurons and the intensity of inhibitory synaptic transmission, facilitating morphine withdrawal responses.
YTHDF1 from Sst-expressing neurons facilitated morphine withdrawal responses
GABAergic cells are the primary source of inhibition in the adult brain, and different subtypes of GABAergic neurons perform certain functions in the brain [37]. Next, we aimed to explore whether different subtypes of inhibitory neurons in the vlPAG exert different effects during morphine withdrawal. The neuropeptide somatostatin (Sst), the calcium-binding protein parvalbumin (PV), and the ionotropic serotonin receptor 5HT3a (5HT3aR) are the three main markers of GABAergic inhibitory neurons [38]. The scRNA-seq data of the Gad2 + inhibitory neurons extracted from the GEO sample GSE240626 [32, 33] were used to explore the expression of the YTHDF1 and NF-κB genes in each inhibitory neuron cluster in the vlPAG, and a total of 4791 cell transcriptomes were retained for subsequent analysis and annotated into 7 major inhibitory neuron clusters (Fig. 7). More information about the normalized expression of cell markers in each cell cluster is shown in Additional file 1: Fig. S5. As shown in Fig. 7, YTHDF1 and NF-κB were expressed in Sst-expressing inhibitory neurons in the vlPAG.

Expression levels of the YTHDF1 or NF-κB genes in different types of inhibitory neurons in the vlPAG based on single-cell RNA-seq data. a UMAP plot of 4791 single cells grouped into 7 major types of inhibitory neurons among the Gad2 + inhibitory neurons in the vlPAG. Each dot represents a single cell, which is colored according to the cell type. b Feature plots of the normalized expression of marker genes in different types of inhibitory neurons in the vlPAG; the depth of color from gray to blue represents low to high expression. c Violin plots of the normalized expression of marker genes in different types of inhibitory neurons in the vlPAG. d Dot plot showing the expression levels of YTHDF1, TNF-α, IL-1β, IL-6, and NF-κB in each inhibitory neuron cluster in the vlPAG. e Feature plot of the normalized expression of the YTHDF1 gene in each inhibitory neuron cluster in the vlPAG. f Violin plot of the normalized expression of the YTHDF1 gene in each inhibitory neuron cluster in the vlPAG. g Feature plot of the normalized expression of the NF-κB gene in each inhibitory neuron cluster in the vlPAG. h Violin plot of the normalized expression of the NF-κB gene in each inhibitory neuron cluster in the vlPAG
We further investigated the relationship between YTHDF1 in Sst-expressing neurons and the level of NF-κB to obtain more information about the pathway involved in the modulation of morphine withdrawal responses. When we knocked down YTHDF1 in vlPAG Sst-expressing neurons in normal mice, the levels of NF-κB and p-NFκB were reduced without affecting the ratio of p-NF-κB to NF-κB (Fig. 8a). The overexpression of YTHDF1 in vlPAG Sst-expressing neurons in normal mice resulted in the upregulation of NF-κB and p-NF-κB without affecting the ratio of p-NF-κB to NF-κB (Fig. 8b). We then knocked down YTHDF1 in vlPAG Sst-expressing neurons in morphine withdrawal model mice, resulting in the downregulation of both NF-κB and p-NFκB and slightly reduced p-NF-κB/NF-κB values (Fig. 8c). This finding was verified through the results of IHC (Fig. 8d–e). Inhibition of YTHDF1 in vlPAG Sst-expressing neurons relieved morphine withdrawal-related somatic signs (Fig. 8f) and aversion (Fig. 8g). We further confirmed that the knockdown of NF-κB in Sst-expressing neurons alleviated the physical symptoms of morphine withdrawal and the aversion response (Additional file 1: Fig. S6). We also selectively knocked down YTHDF1 expression in different inhibitory neurons to compare the outcomes. We found that the knockdown of YTHDF1 in Cck-expressing or PV-expressing neurons did not cause changes in morphine withdrawal-induced responses (Additional file 1: Fig. S6). Again, we tested neuronal and synaptic activity after the inhibition of YTHDF1 in Sst-expressing neurons. After the knockdown of YTHDF1 in Sst-expressing neurons, the co-expression of YTHDF1 and c-fos was significantly decreased (Fig. 9a). We injected AAV-EF1α-DIO-GCaMp6s with Sst-Cre into the vlPAG to detect Ca2+ signals in Sst-expressing neurons, and downregulation of YTHDF1 in Sst-expressing neurons elicited a notable reduction in Ca2+ signals in the vlPAG (Fig. 9b–c), similar to the downregulation of YTHDF1 in inhibitory neurons. The density of dendritic spines in Sst-expressing neurons, marked by Sst-FLP:FDIO-EGFP, was reduced with the downregulation of YTHDF1 in Sst-expressing neurons (Fig. 9d). By recording the mIPSCs of Sst-expressing neurons, we found that the frequency of mIPSCs in the vlPAG was significantly decreased without significant changes in the amplitude of mIPSCs (Fig. 9e–g), indicating that inhibitory synaptic transmission was reduced when Sst-expressing neurons were inhibited. These results indicate that the vlPAG YTHDF1 that participates in modulating morphine withdrawal responses originates from Sst-expressing neurons. Therefore, we assume that the modulation of morphine withdrawal responses is mediated mainly by YTHDF1 in vlPAG Sst-expressing inhibitory neurons by regulating the expression and phosphorylation level of NF-κB and mediating the neuronal and synaptic activities of Sst-expressing inhibitory neurons.

The knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG resulted in the downregulation of NF-κB and p-NF-κB and attenuated morphine withdrawal responses. a Western blotting revealed that the expression of NF-κB was decreased (**P < 0.01, t test, n = 6) after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG of normal mice, without affecting the ratio of p-NF-κB to NF-κB (N.S. = not significant, t test, n = 6). b Western blotting revealed that the expression of NF-κB was increased (**P < 0.01, t test, n = 6) after the overexpression of YTHDF1 in Sst-expressing neurons in the vlPAG of normal mice, but the ratio of p-NF-κB to NF-κB was not significantly different (N.S. = not significant, t test, n = 6). c Western blotting revealed that the expression of NF-κB (**P < 0.01, t test, n = 6) and the ratio of p-NF-κB to NF-κB (*P < 0.05, t test, n = 6) were reduced after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG of morphine withdrawal model mice. d–e IHC images showing the co-expression of NF-κB (d) or p-NF-κB (e) and YTHDF1, revealing reduced NF-κB (d) or p-NF-κB (e) levels after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG. f The morphine withdrawal score (***P < 0.001, t test, n = 10) was decreased after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG. g Aversion was alleviated after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG (**P < 0.01, t test, n = 10). The data are shown as the means ± SEMs

Knockdown of YTHDF1 in Sst-expressing neurons attenuated neuronal activity and inhibited synaptic transmission in the vlPAG. a IHC images showing the reduced co-expression of YTHDF1 in c-fos + neurons after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG (scale bar = 100 μm); the corresponding statistical results are shown (***P < 0.001, t test, n = 6). b–c Ca2+ signaling was reduced in morphine withdrawal model mice after the inhibition of YTHDF1 in vlPAG Sst-expressing neurons, and the corresponding mean GCaMp6s signal, along with the statistical results are shown (***P < 0.001, t test, n = 6). d The density of dendritic spines was significantly reduced after the knockdown of YTHDF1 in Sst-expressing neurons in the vlPAG (**P < 0.01, t test, n = 6). e–g Representative traces of mIPSCs recorded in vlPAG Sst-expressing neurons from morphine withdrawal mice and statistical data for the average frequency and amplitude of mIPSCs showing the decreased mIPSC frequency after the knockdown of YTHDF1 in vlPAG Sst-expressing neurons (**P < 0.01, t test, n = 6). The data are shown as the means ± SEMs
Discussion
In the present study, we showed that YTHDF1 is upregulated in the vlPAG of morphine withdrawal model mice. The knockdown of vlPAG YTHDF1 in inhibitory neurons, but not excitatory neurons, alleviates the morphine withdrawal response. In addition, only YTHDF1 in Sst-expressing neurons, rather than in PV-expressing or Cck-expressing neurons, affects morphine withdrawal responses. Morphine withdrawal causes increased neuronal activity and inhibitory synaptic transmission in mice, which can be reversed by the inhibition of YTHDF1 originating from inhibitory neurons and Sst-expressing neurons in the vlPAG. Downregulation of YTHDF1 leads to decreased levels of NF-κB and p-NF-κB. In addition, the knockdown of NF-κB in Sst-expressing neurons also attenuates the morphine withdrawal responses. The YTHDF1-NF-κB pathway in Sst-expressing neurons in the vlPAG participates in modulating morphine withdrawal responses.
The role of the PAG in the process of morphine withdrawal has been suggested in published studies [39, 40]. Our previous study showed that GABAergic neurons in the vlPAG can modulate morphine withdrawal-induced conditioned place aversion [9]. This finding is consistent with the present study. As the most prevalent internal RNA modification of mammalian mRNAs, m6A has been reported to modulate neuronal functions, including dopaminergic signaling in the mouse midbrain [41]. Upregulation of m6A has been observed with increasing behavioral experience and memory formation [12, 42]. YTHDF1, an m6A-binding protein, can facilitate behavioral experiences and memory processes through its ability to modulate m6A-modified mRNAs [43]. We demonstrated that the levels of YTHDF1 and its relative mRNA expression in the vlPAG are increased in morphine withdrawal model mice. The physical symptoms and CPA induced by morphine withdrawal might be directly related to the upregulation of YTHDF1 in the vlPAG. This result is supported by the finding that not only the somatic signs of morphine withdrawal but also the aversion score improved after the knockdown of YTHDF1 in the vlPAG of WT mice and YTHDF1fl/fl mice. In the present study, the results revealed that YTHDF1 from the inhibitory neurons in the vlPAG affects the morphine withdrawal response, whereas YTHDF1 from the excitatory neurons is unlikely to cause changes in the response. We previously reported that the inhibition of vlPAG GABAergic neurons significantly attenuates the conditioned place aversion (CPA) induced by naloxone-precipitated morphine withdrawal, whereas the activation or inhibition of glutamatergic neurons or the activation of GABAergic neurons does not affect or alter the CPA response [9]. However, Sst-expressing glutamatergic neurons in the lateral/ventrolateral PAG might facilitate mechanical and thermal hypersensitivity and affect behavior in mice with neuropathic pain [44]. The discrepancy between the results of these studies is probably due to the factors being studied and the differences in the neural circuits that mediate morphine withdrawal and neuropathic pain. The midbrain PAG plays a pivotal role in integrating a range of analgesic, behavioral, and autonomic responses to threat, stress, and pain [45]. Opioids and cannabinoids are thought to activate descending analgesic pathways by relieving GABAergic inhibition of the PAG [46, 47]. The inhibitory control of PAG descending outputs is thought to be derived from GABAergic interneurons within this brain structure [16, 48]. Lau et al. reported that opioids activate the descending PAG-RVM analgesic pathway by relieving GABAergic inhibition of PAG neurons, which project to the RVM [49]. Bryony et al. showed that opioids nonselectively disinhibit PAG output neurons and interneurons by acting on both intrinsic and extrinsic GABAergic neurons [50]. When morphine administration is terminated in mice, the effect of relieving the GABAergic inhibition of PAG is weakened, with the upregulation of YTHDF1 expression in inhibitory neurons. Therefore, when YTHDF1 originating from vlPAG inhibitory neurons is knocked down, the inhibitory effects of the inhibitory neurons mediated by YTHDF1 are weakened, which could be one of the important pathways for the modulation of morphine withdrawal responses. Compared with those of normal mice, the number of c-fos + neurons was greater, the calcium signal was stronger in morphine withdrawal model mice, and the density of dendritic spines and the frequency of mIPSCs were greater. These results indicate that the morphine withdrawal response might be related to strengthened neuronal activity and inhibitory synaptic transmission in the vlPAG. The knockdown of YTHDF1 in vlPAG inhibitory neurons and Sst-expressing neurons, which attenuated morphine withdrawal responses, resulted in a decreased number of c-fos + neurons, weakened calcium signals, a lower density of dendritic spines and a lower frequency of mIPSCs, suggesting that vlPAG YTHDF1 might regulate morphine withdrawal responses through the modulation of neuronal and synaptic activities. Our results revealed that, in morphine withdrawal model mice, the frequency of mIPSCs increased, whereas the activity of neurons increased. The increased frequency of mIPSCs suggests increased inhibitory synaptic transmission, probably indicating hyperexcitability in GABAergic neurons in the PAG [51, 52]. We are not sure whether the increased levels of presynaptic inhibitory transmitter could exert an auto-mediating effect on the neurons themselves and further affect neuroexcitability. Moreover, we are not sure whether the release of other transmitters, such as glutamate, are altered, which might further affect the excitability of neurons [53]. The decreased frequency of mIPSCs suggested reduced inhibitory transmitter release. The interaction between the density of dendritic spines and the frequency of mIPSCs in this study requires additional experiments for verification.
In terms of neuron numbers, GABAergic neurons constitute 10 to 20% of the neuronal population across all cortical circuits [54, 55]. The response of subtypes of GABAergic interneurons (INs) to neuromodulators varies, which profoundly affects the function of neocortical circuits and is responsible for the dynamic changes associated with different brain states and behavioral contexts [56]. The characterization of GABAergic neurons involves examining the expression of certain markers, including the calcium-binding protein parvalbumin (PV), the neuropeptide somatostatin (Sst), and the ionotropic serotonin receptor 5HT3a (5HT3aR) [38]. PV, Sst, and 5HT3aR reportedly account for nearly 100% of the GABAergic neurons in the primary somatosensory cortex, and Cck-expressing cells represent an important subgroup of 5HT3aR interneurons. Sst- and PV-INs, which function in the prelimbic cortex as a disinhibitory architecture, are coordinated by morphine via different opioid receptors to disinhibit pyramidal neurons and enhance reward [57]. Kirstie et al. reported that fear conditioning activates a heterogeneous neuronal population in the medial prefrontal cortex that is largely composed of Sst-INs [58]. Researchers have not clearly determined whether morphine withdrawal responses engage specific inhibitory neurons in the vlPAG. Thus, we explored the roles of different inhibitory neuron subtypes in the vlPAG in morphine withdrawal. Our analysis of scRNA-seq data revealed that YTHDF1 is expressed within Sst-expressing inhibitory neurons. Our results confirmed that the inhibition of YTHDF1 in Sst-expressing neurons in the vlPAG effectively alleviated somatic signs of morphine withdrawal and morphine withdrawal-induced aversion. On the other hand, the expression of YTHDF1 in PV-expressing inhibitory neurons or Cck-expressing inhibitory neurons was very low in the scRNA-seq analysis. Our results indicated that the inhibition of YTHDF1 in PV-expressing or Cck-expressing neurons in the vlPAG did not result in changes in morphine withdrawal responses. However, we cannot conclude that the PV-expressing or Cck-expressing neurons in the vlPAG do not participate in the developmental process of morphine withdrawal. These neurons may participate in modulation through other pathways.
We previously reported that D1R-MSNs or D2R-MSNs in the nucleus accumbens (NAc) contribute to the developmental process of morphine withdrawal but play different roles in aspects of behavior or psychology [59]. Zhu et al. observed that excitatory afferents arising from the paraventricular nucleus of the thalamus and basolateral amygdala differentially control D2 and D1 MSNs in the NAc, leading to somatic and emotional abnormalities after withdrawal [60]. In the present study, we showed that the knockdown of YTHDF1 in inhibitory neurons in the vlPAG not only relieves somatic signs of morphine withdrawal but also improves the aversion response. This discrepancy might be due to the different brain regions and circuits studied. The downstream effect of YTHDF1 from inhibitory neurons in the vlPAG remains unclear. Whether the inhibition of YTHDF1 in the vlPAG could simultaneously affect D1R-MSNs and D2R-MSNs in the NAc to alter the somatic signs of morphine withdrawal and aversion is not clear.
We have made efforts to investigate the role of the YTHDF1‒TRAF6 pathway in morphine tolerance and hyperalgesia [14]. We found that IL-1β, IL-6, TNF-α, and NF-κB are regulated by YTHDF1 in the PAG in the previous study [14]. However, only NF-κB, but not IL-1β, IL-6, or TNF-α, interfered with the morphine withdrawal response in the present study. The levels of NF-κB and p-NF-κB were reduced after the knockdown of vlPAG YTHDF1 in morphine withdrawal model mice, whereas the degree of reduction in p-NF-κB levels was greater than that in NF-κB levels. We found that the levels of NF-κB and p-NF-κB were reduced or increased simultaneously with the downregulation or upregulation of YTHDF1 in Sst-expressing neurons in normal mice, respectively, without causing a significant change in the ratio of p-NF-κB to NF-κB. Our speculation is that the level of NF-κB might be modulated by YTHDF1, whereas the level of p-NF-κB is regulated not only by YTHDF1 but also by morphine withdrawal. This finding is consistent with those of previous studies. Russo et al. documented that the inhibition of NF-κB decreases the number of basal dendritic spines on NAc neurons after chronic cocaine exposure and blocks the rewarding effects of cocaine and the ability of previous cocaine exposure to increase the preference for cocaine [61]. The phosphorylation of NF-κB in the NAc has been reported to play a critical role in morphine-induced conditioned place preference [62], and the inhibition of NF-κB has been reported to attenuate morphine-naloxone-induced opioid withdrawal syndrome [63]. In this study, the results of the analysis of the scRNA sequencing data revealed that the expression levels of IL-1β, IL-6, and TNF-α were very low within the vlPAG inhibitory neurons. Proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, are expressed mainly in microglia or astrocytes. In this study, we explored the reactions of these proinflammatory cytokines in inhibitory neurons. Other cytokines, such as IL-1β, IL-6, and TNF-α, might modulate the state of neuroinflammation in mice with morphine withdrawal through other pathways. Whether the inhibitory neurons in the vlPAG directly release inflammatory factors to mediate morphine withdrawal responses remains unclear, and more experiments are needed to confirm these findings.
This study has limitations. Notably, animals treated with saline instead of morphine were commonly used as controls for naloxone-precipitated withdrawal in other studies, which is different from our study design. A previous study showed that the administration of naloxone without chronic morphine exposure does not induce symptoms similar to those induced by morphine withdrawal [60]. A more comprehensive approach for our study would be to include another control group in which morphine was replaced with saline. Moreover, the detailed mechanism by which synaptic activity and electrophysiology affect morphine withdrawal has not been revealed. The exact effect of NF-κB on the modulation of morphine withdrawal and its clear pathway are still uncertain.
Conclusions
In summary, our findings reveal that YTHDF1 originating from inhibitory neurons, especially Sst-expressing neurons in the vlPAG, is crucial for the modulation of morphine withdrawal responses and that the underlying mechanism might be related to the regulation of the expression and phosphorylation of NF-κB. This mechanism may provide a target for the management of opioid withdrawal responses in the population with opioid addiction.
Availability of data and materials
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Abbreviations
- YTHDF1:
-
YTH N6-methyladenosine RNA binding protein 1
- PAG:
-
Periaqueductal gray
- vlPAG:
-
Ventrolateral periaqueductal gray
- m6A:
-
N6-methyladenosine
- mRNA:
-
Messenger RNA
- GABA:
-
γ-Aminobutyric acid
- CPA:
-
Conditioned place aversion
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- TNF-α:
-
Tumor necrosis factor-α
- NF-κB:
-
Nuclear factor-κB
- YTHDF1fl/fl :
-
YTHDF1flox/flox
- WT:
-
Wild-type
- PV:
-
Parvalbumin
- Cck:
-
Cholecystokinin
- Sst:
-
Somatostatin
References
Volkow ND, Jones EB, Einstein EB, Wargo EM. Prevention and treatment of opioid misuse and addiction. JAMA Psychiatry. 2019;76(2):208.
Stinus L, Caille S, Koob GF. Opiate withdrawal-induced place aversion lasts for up to 16 weeks. Psychopharmacology. 2000;149(2):115–20.
Behbehani MM. Functional characteristics of the midbrain periaqueductal gray. Prog Neurobiol. 1995;46(6):575–605.
Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5(7):565–75.
Motta SC, Carobrez AP, Canteras NS. The periaqueductal gray and primal emotional processing critical to influence complex defensive responses, fear learning and reward seeking. Neurosci Biobehav Rev. 2017;76:39–47.
Bozarth MA. Physical dependence produced by central morphine infusions: an anatomical mapping study. Neurosci Biobehav Rev. 1994;18(3):373–83.
Yi H, Iida T, Liu S, Ikegami D, Liu Q, Iida A, et al. IL-4 mediated by HSV vector suppresses morphine withdrawal response and decreases TNFα, NR2B, and pC/EBPβ in the periaqueductal gray in rats. Gene Ther. 2017;24(4):224–33.
Hao S, Liu S, Zheng X, Zheng W, Ouyang H, Mata M, et al. The role of TNFα in the periaqueductal gray during naloxone-precipitated morphine withdrawal in rats. Neuropsychopharmacology. 2010;36(3):664–76.
Bai X, Zhang K, Ou C, Nie B, Zhang J, Huang Y, et al. Selective activation of AKAP150/TRPV1 in ventrolateral periaqueductal gray GABAergic neurons facilitates conditioned place aversion in male mice. Commun Biol. 2023;6(1):742.
Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540(7632):242–7.
Weng Y-L, Wang X, An R, Cassin J, Vissers C, Liu Y, et al. Epitranscriptomic m6A regulation of axon regeneration in the adult mammalian nervous system. Neuron. 2018;97(2):313-25.e6.
Widagdo J, Zhao Q-Y, Kempen M-J, Tan MC, Ratnu VS, Wei W, et al. Experience-dependent accumulation of N6-methyladenosine in the prefrontal cortex is associated with memory processes in mice. J Neurosci. 2016;36(25):6771–7.
Shi H, Zhang X, Weng Y-L, Lu Z, Liu Y, Lu Z, et al. m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature. 2018;563(7730):249–53.
Ouyang H, Zhang J, Chi D, Zhang K, Huang Y, Huang J, et al. The YTHDF1–TRAF6 pathway regulates the neuroinflammatory response and contributes to morphine tolerance and hyperalgesia in the periaqueductal gray. J Neuroinflamm. 2022;19(1):310.
Benarroch EE. Periaqueductal gray. Neurology. 2012;78(3):210–7.
Samineni VK, Grajales-Reyes JG, Copits BA, O’Brien DE, Trigg SL, Gomez AM, et al. Divergent modulation of nociception by glutamatergic and GABAergic neuronal subpopulations in the periaqueductal gray. Eneuro. 2017;4(2):ENEURO.0129-16.2017.
Gao ZR, Chen WZ, Liu MZ, Chen XJ, Wan L, Zhang XY, et al. Tac1-expressing neurons in the periaqueductal gray facilitate the itch-scratching cycle via descending regulation. Neuron. 2019;101(1):45-59.e9.
Eidson LN, Inoue K, Young LJ, Tansey MG, Murphy AZ. Toll-like receptor 4 mediates morphine-induced neuroinflammation and tolerance via soluble tumor necrosis factor signaling. Neuropsychopharmacology. 2016;42(3):661–70.
Wang H, Huang M, Wang W, Zhang Y, Ma X, Luo L, et al. Microglial TLR4-induced TAK1 phosphorylation and NLRP3 activation mediates neuroinflammation and contributes to chronic morphine-induced antinociceptive tolerance. Pharmacolog Res. 2021;165:105482.
Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115(1):50–9.
Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22(22):9980–9.
Bai L, Zhai C, Han K, Li Z, Qian J, Jing Y, et al. Toll-like receptor 4-mediated nuclear factor-κB activation in spinal cord contributes to chronic morphine-induced analgesic tolerance and hyperalgesia in rats. Neurosci Bull. 2014;30(6):936–48.
Zeng X, Lin MY, Wang D, Zhang Y, Hong Y. Involvement of adrenomedullin in spinal glial activation following chronic administration of morphine in rats. Eur J Pain. 2014;18(9):1323–32.
Doyle TM, Hutchinson MR, Braden K, Janes K, Staikopoulos V, Chen Z, et al. Sphingosine-1-phosphate receptor subtype 1 activation in the central nervous system contributes to morphine withdrawal in rodents. J Neuroinflamm. 2020;17(1):314.
Quezada M, Ponce C, Berríos-Cárcamo P, Santapau D, Gallardo J, De Gregorio C, et al. Amelioration of morphine withdrawal syndrome by systemic and intranasal administration of mesenchymal stem cell-derived secretome in preclinical models of morphine dependence. CNS Neurosci Ther. 2023;30:e14517.
Gonek M, McLane VD, Stevens DL, Lippold K, Akbarali HI, Knapp PE, et al. CCR5 mediates HIV-1 Tat-induced neuroinflammation and influences morphine tolerance, dependence, and reward. Brain Behav Immun. 2018;69:124–38.
Li T, Tan Y-T, Chen Y-X, Zheng X-J, Wang W, Liao K, et al. Methionine deficiency facilitates antitumour immunity by altering m6A methylation of immune checkpoint transcripts. Gut. 2023;72(3):501–11.
Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, et al. Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science. 1996;273(5275):657–9.
Zachariou V, Brunzell DH, Hawes J, Stedman DR, Bartfai T, Steiner RA, et al. The neuropeptide galanin modulates behavioral and neurochemical signs of opiate withdrawal. Proc Natl Acad Sci. 2003;100(15):9028–33.
Papaleo F, Contarino A. Gender- and morphine dose-linked expression of spontaneous somatic opiate withdrawal in mice. Behav Brain Res. 2006;170(1):110–8.
Cruz HG, Berton F, Sollini M, Blanchet C, Pravetoni M, Wickman K, et al. Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J Neurosci. 2008;28(15):4069–77.
Wang X, Wu X, Wu H, Xiao H, Hao S, Wang B, et al. Neural adaption in midbrain GABAergic cells contributes to high-fat diet–induced obesity. Sci Adv. 2023;9(44):eadh2884.
Wu H, Wang X. Neural adaption in midbrain GABAergic cells contributes to high-fat-diet induced obesity. GEO. 2023. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE240626.
McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8(4):329-37.e4.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888-902.e21.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–96.
Caputi A, Melzer S, Michael M, Monyer H. The long and short of GABAergic neurons. Curr Opin Neurobiol. 2013;23(2):179–86.
Lee S, Hjerling-Leffler J, Zagha E, Fishell G, Rudy B. The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci. 2010;30(50):16796–808.
Vázquez-León P, Miranda-Páez A, Chávez-Reyes J, Allende G, Barragán-Iglesias P, Marichal-Cancino BA. The periaqueductal gray and its extended participation in drug addiction phenomena. Neurosci Bull. 2021;37(10):1493–509.
Iida T, Yi H, Liu S, Ikegami D, Zheng W, Liu Q, et al. MnSOD mediated by HSV vectors in the periaqueductal gray suppresses morphine withdrawal in rats. Gene Ther. 2017;24(5):314–24.
Hess ME, Hess S, Meyer KD, Verhagen LAW, Koch L, Brönneke HS, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16(8):1042–8.
Walters BJ, Mercaldo V, Gillon CJ, Yip M, Neve RL, Boyce FM, et al. The role of the RNA demethylase FTO (fat mass and obesity-associated) and mRNA methylation in hippocampal memory formation. Neuropsychopharmacology. 2017;42(7):1502–10.
Wang X, Zhao Boxuan S, Roundtree Ian A, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Zhang Y, Huang X, Xin W-J, He S, Deng J, Ruan X. Somatostatin neurons from periaqueductal gray to medulla facilitate neuropathic pain in male mice. J Pain. 2023;24(6):1020–9.
Keay KA, Bandler R. Parallel circuits mediating distinct emotional coping reactions to different types of stress. Neurosci Biobehav Rev. 2001;25(7–8):669–78.
Moreau J-L, Fields HL. Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission. Brain Res. 1986;397(1):37–46.
Depaulis A, Morgan MM, Liebeskind JC. GABAergic modulation of the analgesic effects of morphine microinjected in the ventral periaqueductal gray matter of the rat. Brain Res. 1987;436(2):223–8.
Park C, Kim J-H, Yoon B-E, Choi E-J, Lee CJ, Shin H-S. T-type channels control the opioidergic descending analgesia at the low threshold-spiking GABAergic neurons in the periaqueductal gray. Proc Natl Acad Sci. 2010;107(33):14857–62.
Lau BK, Winters BL, Vaughan CW. Opioid presynaptic disinhibition of the midbrain periaqueductal grey descending analgesic pathway. Br J Pharmacol. 2020;177(10):2320–32.
Winters BL, Lau BK, Vaughan CW. Cannabinoids and opioids differentially target extrinsic and intrinsic GABAergic inputs onto the periaqueductal grey descending pathway. J Neurosci. 2022;42:7744.
Bajo M, Madamba SG, Roberto M, Siggins GR. Acute morphine alters GABAergic transmission in the central amygdala during naloxone-precipitated morphine withdrawal: role of cyclic AMP. Front Integr Neurosci. 2014;8:45.
Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology. 2003;45(5):575–84.
Chen M, Zhao Y, Yang H, Luan W, Song J, Cui D, et al. Morphine disinhibits glutamatergic input to VTA dopamine neurons and promotes dopamine neuron excitation. eLife. 2015;4:e09275.
Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1998;6(4):347–470.
Hu H, Gan J, Jonas P. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345(6196):1255263.
Tremblay R, Lee S, Rudy B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron. 2016;91(2):260–92.
Jiang C, Wang X, Le Q, Liu P, Liu C, Wang Z, et al. Morphine coordinates SST and PV interneurons in the prelimbic cortex to disinhibit pyramidal neurons and enhance reward. Mol Psychiatry. 2019;26(4):1178–93.
Cummings KA, Bayshtok S, Dong TN, Kenny PJ, Clem RL. Control of fear by discrete prefrontal GABAergic populations encoding valence-specific information. Neuron. 2022;110(18):3036-52.e5.
Bai X, Zhang K, Ou C, Mu Y, Chi D, Zhang J, et al. AKAP150 from nucleus accumbens dopamine D1 and D2 receptor-expressing medium spiny neurons regulates morphine withdrawal. iScience. 2023;26(11):26.
Zhu Y, Wang K, Ma T, Ji Y, Lou Y, Fu X, et al. Nucleus accumbens D1/D2 circuits control opioid withdrawal symptoms in mice. J Clin Invest. 2023;133(18):e163266.
Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, et al. Nuclear factor κB signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29(11):3529–37.
Zhang X, Cui Y, Jing J, Cui Y, Xin W, Liu X. Involvement of p38/NF-κB signaling pathway in the nucleus accumbens in the rewarding effects of morphine in rats. Behav Brain Res. 2011;218(1):184–9.
Rehni AK, Bhateja P, Singh TG, Singh N. Nuclear factor-κ-B inhibitor modulates the development of opioid dependence in a mouse model of naloxone-induced opioid withdrawal syndrome. Behav Pharmacol. 2008;19(3):265–9.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (82071237) and the Guangdong Basic and Applied Basic Research Foundation (2023A1515010147).
Author information
Authors and Affiliations
Contributions
OUYHD and ZWA designed the experiments. OCP, ZK, MYY, HZZ, WY and LXL performed the experiments and collected the data. MYY, WY, HW and OCP performed the analysis. OCP, ZK and OUYHD wrote the manuscript. OUYHD and ZWA supervised this study. All the authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experimental procedures were approved by the Use Committee of Sun Yat-sen University and the Animal Care Committee (No. L102012020000X) and were conducted in accordance with the guidelines of the National Institutes of Health (NIH).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12916_2024_3634_MOESM1_ESM.zip
Additional file 1: Figure S1-S7. Figure S1. Supplemental information on calcium signaling and virus efficiency. (a) Schematic of the method for expressing GCaMp6s. (b) Co-staining experiment of Sst-Cre + DIO-EGFP and an antibody against somatostatin showing the specificity of Sst-Cre as a promoter. (c-f) Knockdown efficiency of the CMV-YTHDF1-shRNA (***P < 0.001, t test, n = 6), CamkIIα-YTHDF1-shRNA (**P < 0.01, t test, n = 6), Vgat-YTHDF1-shRNA (***P < 0.001, t test, n = 6), and Sst-YTHDF1-shRNA (**P < 0.01, t test, n = 6). Figure S2. The normalized expression of cell markers in each cell cluster in the PAG. (a) Feature plots showing the normalized expression of cell markers in each cell cluster in the PAG. (b) Violin plots showing the normalized expression of cell markers (rows) in each cell cluster (columns) in the PAG. Cell clusters and the expression level of each gene are indicated on the x- and y-axes, respectively. Figure S3. Expression of YTHDF1 in certain types of neurons. (a) IHC images showing the expression of YTHDF1 in NeuN + neurons. (b) IHC images showing the expression of YTHDF1 in CX3CR1 + neurons. (c) IHC images showing the expression of YTHDF1 in GFAP + neurons. Figure S4. Expression levels of IL-1β, IL-6, and TNF-α after the knockdown of YTHDF1 in the vlPAG. (a-b) IHC images showing that the expression level of IL-1β was not significantly changed after the knockdown of YTHDF1 in the vlPAG (a), which was verified by the western blot results (N.S. = not significant, t test, n = 6) (b). (c-d) IHC images showing that the expression level of IL-6 was not significantly changed after the knockdown of YTHDF1 in the vlPAG (c), which was verified by the western blot results (N.S. = not significant, t test; n = 6) (d). (e–f) IHC images showing that the expression level of TNF-α was not significantly changed after the knockdown of YTHDF1 in the vlPAG (e), which was verified by the western blot results (N.S. = not significant, t test, n = 6) (f). Figure S5. The normalized expression of cellular markers of inhibitory neurons in each cell cluster in the vlPAG. (a) Feature plots showing the normalized expression of cellular markers of inhibitory neurons in each cell cluster in the vlPAG. (b) Violin plots showing the normalized expression of cellular markers of inhibitory neurons (rows) in each cell cluster (columns) in the vlPAG. Cell clusters and the expression level of each gene are indicated on the x- and y-axes, respectively. Figure S6. Supplemental information on the changes in morphine withdrawal responses under different conditions. (a) Morphine withdrawal scores (***P < 0.001, t test, n = 10) were decreased after NF-κB was knocked down in the Sst-expressing inhibitory neurons in the vlPAG. (b) Aversion was alleviated after the knockdown of NF-κB in Sst-expressing inhibitory neurons in the vlPAG (**P < 0.01, t test, n = 10). (c-d) Feature plots of the normalized expression of the marker genes Pvalb (c) or CCK (d) in different inhibitory neuron clusters in the vlPAG. (e) Morphine withdrawal scores were not significantly affected after the knockdown of YTHDF1 in Pvalb-expressing inhibitory neurons in the vlPAG (N.S. = not significant, t test, n = 10). (f) The aversion score did not change significantly after the knockdown of YTHDF1 in Pvalb-expressing inhibitory neurons in the vlPAG (N.S. = not significant, t test, n = 10). (g) The morphine withdrawal score was not significantly affected after the knockdown of YTHDF1 in the CCK-8-expressing inhibitory neurons in the vlPAG (N.S. = not significant, t test, n = 10). (h) The aversion score did not change significantly after the knockdown of YTHDF1 in CCK-expressing inhibitory neurons in the vlPAG (N.S. = not significant, t test, n = 10). Figure S7. Raw images of all western blots analysed in the present study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ou, C., Zhang, K., Mu, Y. et al. YTHDF1 in periaqueductal gray inhibitory neurons contributes to morphine withdrawal responses in mice. BMC Med 22, 406 (2024). https://doi.org/10.1186/s12916-024-03634-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-024-03634-2