
Abstract
Background
Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is an autosomal recessive disorder and one of the most common inherent causes of cholestatic jaundice in Asian infants. Mutations in the SLC25A13 gene, which encodes citrin protein expressed in the liver, have been identified as the genetic cause for NICCD.
Case presentation
Here, we report a 4-month-old female with clinical features including jaundice, hyperbilirubinemia, hyperlactacidemia, and abnormal liver function. The patient was diagnosed with NICCD by differential diagnosis using genetic analysis. Mutations in 60 jaundice-related genes were tested by using amplicon sequencing, which was performed on an Ion S5XL genetic analyzer. A compound heterozygous mutation in the SLC25A13 gene was identified, consisting of a known deletion SLC25A13:c.852_855delTATG and a novel splicing mutation SLC25A13:c.1841 + 3_1841 + 4delAA. Sanger sequencing for the proband and her parents was performed to validate the result and reveal the source of mutations.
Conclusion
A compound heterozygous mutation in the SLC25A13 gene was identified in a 4-month-old female patient with NICCD. Our data suggest that amplicon sequencing is a helpful tool for the differential diagnosis of inherited diseases with similar symptoms. Further studies of the mutation spectrum of neonatal jaundice in China are warranted.
Similar content being viewed by others

Background
Citrin deficiency is an autosomal recessive disorder caused by mutations of the SLC25A13 gene on chromosome 7q21.3. The SLC25A13 gene is mainly expressed in the liver and encodes an aspartate glutamate carrier (AGC), which plays an important role in malate–aspartate NADH shuttling and urea synthesis [1]. There are two major clinical phenotypes of citrin deficiency, adult-onset type II citrullinemia (CTLN2, OMIM: 603471) and neonatal intrahepatic cholestatic hepatitis (NICCD, OMIM: 605814) [2, 3]. Patients with CTLN2 suffer from recurring neuropsychiatric symptoms, such as disorientation, delirium, and delusion, which are generally associated with hyperammonemia. Symptoms of NICCD patients include cholestatic jaundice, hypoproteinemia, hypoglycemia, liver enlargement and dysfunction, most of which usually disappear by 12 months of age without special treatment [4, 5]. However, some patients might develop CTLN2 decades later [6]. In addition, another phenotype different from NICCD and CTLN2 was reported in older child patients, which was referred to as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) [7].
In China, the frequency of carriers with mutations in the SLC25A13 gene is very high and presents significant geographic differences. The frequency was estimated to be 1/940 and 1/48 in northern and southern China, respectively [8]. Recently, the mutation spectrum of the SLC25A13 gene was investigated, resulting in the variations c.851_854del4, c.1638_1660dup, IVS6 + 5G > A and IVS16ins3Kb constituting the high-frequency mutations on top of the list [9]. Molecular diagnosis provides essential evidence for diagnosing citrin deficiency.
In this case, we report a 4-month-old female patient diagnosed with NICCD using amplicon sequencing of whole exons of 60 cholestatic jaundice-related genes. A novel splicing mutation, SLC25A13:c.1841 + 3_1841 + 4delAA, was identified in this patient, compound with a known deletion c.852_855delTATG. The two mutations were confirmed and proven to be separately inherited from father and mother by Sanger sequencing. Our data expand the mutation spectrum of the SLC25A13 gene in Chinese patients with NICCD and demonstrate that amplicon sequencing is accurate and efficient for identifying mutations in patients with inherited diseases.
Case presentation
This study was approved by the ethics committee of the Third Affiliated Hospital of Zhengzhou University. Informed consent from the parents was obtained before collecting blood samples.
A 4-month-old female patient was admitted to the Department of Pediatrics in the Third Affiliated Hospital of Zhengzhou University Hospital for mild neonatal jaundice. The chief complaint was jaundice, coughing and runny nose. Physical examination was normal, and ultrasonic examination showed a slight gallbladder wall thickening. Biliary atresia was excluded based on the normal result of magnetic resonance cholangiopancreatography (MRCP). Blood and liver function tests were performed in the clinical laboratory, and hyperbilirubinemia, hyperlactacidemia, and abnormal liver function were revealed, suggesting neonatal hepatitis in the patient (Table 1). Symptoms were significantly improved after 5 days of treatment, and the patient was discharged with recommendations for diet management and regular examination.
Differential diagnosis for NICCD
The patient was diagnosed with neonatal hepatitis based on neonatal jaundice, hepatomegaly and liver dysfunction. Further differential diagnosis should consider three major pathogenesis, including infection, congenital biliary atresia and inherited metabolic diseases of the liver. CMV and TORCH test were negative, excluding the infectious hepatitis. The level of total bile acid (TBC) was out of normal range, suggesting the possibility of congenital biliary atresia. However, Kaolin stool wasn’t observed, MRCP was normal, which means biliary atresia was also excluded. The likely possible pathogenesis could be inherited metabolic diseases of the liver. In a retrospective study of liver function and islet beta cell functions for 36 patients with NICCD and 50 control individuals indicates that significantly higher of alpha-fetoprotein is one of the typical symptoms in NICCD patients, which is the result of liver dysfunction correlated with islet beta cell functions [10]. Combining the high frequency in Southern China, NICCD is highly suspected [9]. Considering the similar symptoms and complex causative mutations for inherited metabolic liver diseases, high-throughput sequencing provides an effective method of further differential diagnosis on genetic level.
A genetic test was performed to detect potential pathogenic mutations in this patient using high-throughput amplicon sequencing. In brief, genomic DNA was extracted from a 0.5 mL peripheral blood sample, and a custom-designed amplicon sequencing panel purchased from Thermo Fisher Scientific was used to amplify whole exons of 60 cholestatic jaundice-related genes (Additional file 1: Table S1). PCR products were processed and sequenced with the Ion Chef and Ion GeneStudio S5 System, and variants were detected and analyzed using VariantCaller V5.0. A filter was applied to exclude common variants with minor allele frequency (MAF) > 1% in the UCSC, dbSNP and 1000 Genomes Project databases. The clinical significance of the remaining candidate variants was predicted by PolyPhen-2 [11], SIFT [12] and HSF [13] (Additional file 2: Figure S1), the change of splicing site was predicted by NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html) [14].
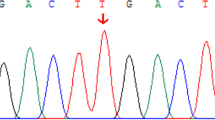
As result, a compound heterozygote in the SLC25A13 gene was identified and considered to be causative for this patient. The frame-shift deletion SLC25A13:c.852_855delTATG (NM_014251.2:c.852_855delTATG) is a pathogenic SNP (rs80338720) with a MAF = 0.0003, according to the gnomAD database. This deletion causes an amino acid sequence change in Met285Profs, which could significantly affect the function of the citrin protein. Another variant in this compound heterozygote is SLC25A13:c.1841 + 3_1841 + 4delAA, which is also referred to as NC_000007.13:g.95750963_95750964delTT. This variant is located in the intron between exon17 and exon18 of the SLC25A13 gene, and it is predicted to be an alteration that most likely affects splicing. Scores predicted by both HSF Matrices and MaxEnt indicate a dramatic change caused by this mutation, probably lead to broken of the wild-type splice-site (Additional file 3: Table S2 and Additional file 4: Figure S2). A new donor site was detected 65 bp downstream, by using the NNSPLICE (Fig. 1c). This splicing mutation is reported for the first time in patient of NICCD.

Mutation detection and splicing site prediction in the SLC25A13 gene. a Identification of frame-shift deletion c.852_855delTATG using Sanger sequencing; b Identification of splice-site mutation c.1841 + 3_1841 + 4delAA using Sanger sequencing; c Change of splice-site predicted by NNSPLICE
To validate the result of amplicon sequencing and investigate the source of these two mutations, we analyzed these two variants for the proband and her parents using Sanger sequencing. The result shows a typical autosomal recessive inheritance pattern in this case. The c.852_855delTATG and c.1841 + 3_1841 + 4delAA mutations were inherited from the patient’s father and mother, respectively (Fig. 1a and b). Both parents are healthy, and no family history was revealed.
Discussion and conclusions
NICCD is one of the most common inherent causes of cholestatic jaundice in Asian infants. In this case, we reported a 4-month-old female patient diagnosed with NICCD by genetic test. The patient was admitted for mild neonatal jaundice. Clinical features and results of laboratory tests suggested neonatal intrahepatic cholestasis. Considering its high frequency in south China [9], NICCD was highly suspected. However, it’s unlikely to give a diagnosis since plasma free amino acid analysis wasn’t a routine test in our clinical laboratory. To identify the pathogenic mutation in this patient, we used a custom-designed amplicon sequencing panel which aiming to make differential diagnosis for pathologic jaundice. As result, a compound heterozygote in the SLC25A13 gene was identified in this patient, which was suggested to be causative by functional prediction. Sanger sequencing confirmed the result and revealed that two heterozygous mutations were inherited from the father and mother, which perfectly match the autosomal recessive inheritance model. The patient was discharged after 5 days of treatment, including anti-infection, liver protection, and cholagogue. Prognosis is good in regular reexamination in the following 2 months.
The frame-shift deletion SLC25A13:c.852_855delTATG detected in this patient is relatively rare, with MAF = 0.0003 in gnomAD, causing an amino acid sequence change in Met285Profs. It had been reported to be pathogenic in several cases in the ClinVar database. There is only one nucleotide difference between this mutation and the most frequent variation c.851_854del4; however, the prevalence of these two mutations in patients with citrin deficiency varies greatly. The mutation c.1841 + 3_1841 + 4delAA is reported for the first time, which is predicted to affect RNA splicing, resulting a new donor site downstream. Splice-site mutations are rare in the SLC25A13 mutation spectrum, representing nearly 1/10 (4 in 41) of mutations composing the spectrum [9]. In some inherited diseases, symptoms with different severities could be caused by different genotypes [15]; however, the genotype-phenotype correlations of citrin deficiency remain unknown. A previous study had investigated these correlations initially, but no clear conclusion was established [16]. Obviously, large cohort investigations and genetic screening are required. In this case, we identified mutations within 24 h using amplicon sequencing, and the result was perfectly matched with Sanger sequencing. This indicates that amplicon sequencing is helpful for diagnosis and further studies of inherited diseases.
In summary, we identified a compound heterozygous mutation in the SLC25A13 gene from a patient with citrin deficiency, consisting of a known deletion (c.852_855delTATG) and a novel splicing mutation (c.1841 + 3_1841 + 4delAA). Both mutations were predicted to be harmful for protein function. Pedigree analysis was performed using Sanger sequencing, demonstrating that the mutations were inherited from the patient’s father and mother. For patients with neonatal jaundice, accurate and efficient differential diagnosis is important for appropriate treatment and prognosis. Our data suggest that amplicon sequencing could play a useful role in the clinical diagnosis of inherited diseases.
Availability of data and materials
Not applicable.
Abbreviations
- AGC:
-
Aspartate glutamate carrier
- CTLN2:
-
Citrin deficiency, adult-onset type II citrullinemia
- MAF:
-
Minor allele frequency
- MRCP:
-
Magnetic resonance cholangiopancreatography
- NICCD:
-
neonatal intrahepatic cholestasis caused by citrin deficiency
References
Sinasac DS, Crackower MA, Lee JR, Kobayashi K, Saheki T, Scherer SW, Tsui L. Genomic structure of the adult-onset type II citrullinemia gene, SLC25A13, and cloning and expression of its mouse homologue. Genomics. 1999;62(2):289–92.
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999;22(2):159–63.
Tazawa Y, Kobayashi K, Ohura T, Abukawa D, Nishinomiya F, Hosoda Y, Yamashita M, Nagata I, Kono Y, Yasuda T. Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. J Pediatr. 2001;138(5):735–40.
Tazawa Y, Kobayashi K, Abukawa D, Nagata I, Maisawa S, Sumazaki R, Iizuka T, Hosoda Y, Okamoto M, Murakami J. Clinical heterogeneity of neonatal intrahepatic cholestasis caused by citrin deficiency: case reports from 16 patients. Mol Genet Metab. 2004;83(3):213–9.
Ohura T, Kobayashi K, Tazawa Y, Abukawa D, Sakamoto O, Tsuchiya S, Saheki T. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis. 2007;30(2):139–44.
Tomomasa T, Kobayashi K, Kaneko H, Shimura H, Fukusato T, Tabata M, Inoue Y, Ohwada S, Kasahara M, Morishita Y. Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy. J Pediatr. 2001;138(5):741–3.
Song Y, Guo L, Yang Y, Han LS, Kobayashi K, Saheki T. Failure to thrive and dyslipidemia caused by citrin deficiency: a novel clinical phenotype. Chinese J Contemp Pediatr. 2009;11(5):328.
Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet. 2005;50(7):338–46.
Lin W, Zeng H, Zhang Z, Mao M, Zheng Q, Zhao S, Cheng Y, Chen F, Wen W, Song Y. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci Rep. 2016;6(1):29732.
Lu C-T, Yang J, Huang S-M, Feng L, Li Z-J. Analysis of islet beta cell functions and their correlations with liver dysfunction in patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). Medicine. 2017;96(45):e8638.
Adzhubei I, Schmidt S, Peshkin L, Ramensky V, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Sim N, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:452–7.
Desmet F, Hamroun D, Lalande M, Collodberoud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol. 1997;4(3):311–23.
Westbroek W, Adams DR, Huizing M, Koshoffer A, Dorward H, Tinloy B, Parkes J, Helipwooley A, Kleta R, Tsilou E. Cellular defects in Chediak–Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Investig Dermatol. 2007;127(11):2674–7.
Devi ARR, Naushad SM. SLC25A13 c.1610_1612delinsAT mutation in an Indian patient and literature review of 79 cases of citrin deficiency for genotype-phenotype associations. Gene. 2018;668:190–5.
Acknowledgments
The authors gratefully appreciate the patient and her family for their participation in this clinical study. This work was supported by the Third Affiliated Hospital of Zhengzhou University (Children’s Hospital Affiliated to Zhengzhou University).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
LZ and YYL performed DNA extraction and amplicon sequencing, analyzed the sequencing data, and wrote the manuscript. WS, JG and YT carried out clinical examination and Sanger sequencing. YL and YG revised the manuscript. SC designed this study and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics committee of the Third Affiliated Hospital of Zhengzhou University. Informed consent for publishing data and images was obtained from the parents before performing the genetic test.
Consent for publication
A copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Details of Amplicon sequencing panel for 60 cholestatic jaundice related genes.
Additional file 2: Figure S1.
Workflow of detecting causative mutations for inherited disease.
Additional file 3:
Table S2. Result of splice-site prediction.
Additional file 4: Figure S2.
Result of functional prediction for SLC25A13:c.1841+3_1841+4delAA by using HSF.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Zhang, L., Li, Y., Shi, W. et al. Identification of a novel splicing mutation in the SLC25A13 gene from a patient with NICCD: a case report. BMC Pediatr 19, 348 (2019). https://doi.org/10.1186/s12887-019-1751-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1751-9