
Abstract
Background
The cytoplasmic male sterility (CMS) of rice is caused by chimeric mitochondrial DNA (mtDNA) that is maternally inherited in the majority of multicellular organisms. Wild rice (Oryza rufipogon Griff.) has been regarded as the ancestral progenitor of Asian cultivated rice (Oryza sativa L.). To investigate the distribution of original CMS source, and explore the origin of gametophytic CMS gene, a total of 427 individuals with seventeen representative populations of O. rufipogon were collected in from Dongxiang of Jiangxi Province to Sanya of Hainan Province, China, for the PCR amplification of atp6, orfH79 and B-atp6-orfH79, respectively.
Results
The B-atp6-orfH79 and its variants (B-atp6-GSV) were detected in five among seventeen populations (i.e. HK, GZ, PS, TL and YJ) through PCR amplification, which could be divided into three haplotypes, i.e., BH1, BH2, and BH3. The BH2 haplotype was identical to B-atp6-orfH79, while the BH1 and BH3 were the novel haplotypes of B-atp6-GSV. Combined with the high-homology sequences in GenBank, a total of eighteen haplotypes have been revealed, only with ten haplotypes in orfH79 and its variants (GSV) that belong to three species (i.e. O. rufipogon, Oryza nivara and Oryza sativa). Enough haplotypes clearly demonstrated the uniform structural characteristics of the B-atp6-orfH79 as follows: except for the conserved sequence (671 bp) composed of B-atp6 (619 bp) and the downstream followed the B-atp6 (52 bp, DS), and GSV sequence, a rich variable sequence (VS, 176 bp) lies between the DS and GSV with five insertion or deletion and more than 30 single nucleotide polymorphism. Maximum likelihood analysis showed that eighteen haplotypes formed three clades with high support rate. The hierarchical analysis of molecular variance (AMOVA) indicated the occurrence of variation among all populations (FST = 1; P < 0.001), which implied that the chimeric structure occurred independently. Three haplotypes (i.e., H1, H2 and H3) were detected by the primer of orfH79, which were identical to the GVS in B-atp6-GVS structure, respectively. All seventeen haplotypes of the orfH79, belonged to six species based on our results and the existing references. Seven existed single nucleotide polymorphism in GSV section can be translated into eleven various amino acid sequences.
Conclusions
Generally, this study, indicating that orfH79 was always accompanied by the B-atp6, not only provide two original CMS sources for rice breeding, but also confirm the uniform structure of B-atp-orfH79, which contribute to revealing the origin of rice gametophytic CMS genes, and the reason about frequent recombination of mitochondrial DNA.
Similar content being viewed by others

Background
Cytoplasmic male sterility (CMS) caused by orfH79 and its variants has been used to produce rice hybrid seeds of several types gametophytic CMS lines on a commercial scale, which eliminates the need for hand emasculation [1,2,3,4]. The CMS genes are located in the mitochondrial genome [5,6,7,8,9,10,11,12], and maternally inherited in the majority of angiosperms [13, 14]. Given its maternal inheritance, the original CMS source can be bred into the CMS line through the successive back-crossing as the female parent for the selection of different isoplasmic allonuclear male-sterile plant [15]. Plant mitochondrial DNA (mtDNA) is highly conserved as a consequence of its maternal inheritance. And its notoriously complex structure [14, 16] reflects in large part the frequent occurrence of recombination, integration, and deletion of DNA sequences [17, 18]. Almost all reported CMS genes are chimeric structure, i.e., the exogenous sequence in the gene originated from nuclear DNA, chloroplast DNA, or mtDNA itself [19]. Double-strand break repair is one hypothesized mechanism for mtDNA recombination [20]. Cell-to-cell movement of mitochondria through a stem graft junction involving two tobacco (Nicotiana) species has been reported [21]. If the problem of CMS is regarded as a tree, the application of the sterile gene is the crown, the action principle of the sterile gene is the trunk, while the origin of the sterile gene is the root. At present, the studies about the first two are too numerous to list, but the latter is too few. However, the mechanism by which chimeric CMS genes arose and resulted in sterility with such consistency is uncertain.
The generation of CMS system mainly attributed to the three phenomena as follows: (1) spontaneous sterility, such as rice Wild Abortive type CMS (CMS-WA) [22], Polish rape CMS-POL, and radish CMS-Ogura [23]; (2) distant hybridization, such as rice Boro II type CMS (CMS-BT) [24], HongLian type CMS (CMS-HL) [22], and sorghum CMS-IS1112C (A3) [25]; and (3) somatic hybridization [26]. Among them, distant hybridization is the most common means of generating CMS system. For instance, distant hybridization is responsible for more than sixty CMS original sources from rice in China [27], and eighty-four CMS original sources from corn in the USA [28]. Additionally, the interspecific hybridization also generated seventy-two CMS sources in sunflower [29]. Transgene can also produce male sterility, which have not been applied in the production [30].
Generally, some CMS systems in crops are considered to have originated in their closely related wild species with sterility gene. A survey of the CMS gene orfH522 distribution in sunflower, using a gene-specific primer among more than 1,200 plants representing fifty-five accessions of Helianthus annuus and twenty-six accessions of Helianthus petiolaris, failed to detect CMS cytotypes in natural populations [31]. The orf138 gene responsible for Ogura CMS, arose only once in the evolution of radish as indicated by a survey of wild and cultivated radish accessions that were grown or distributed where the Ogura cytoplasm was originally identified in Japan [32]. The detection of orf138 gene in natural populations of Raphanus raphanistrum remained at a low frequency in Europe, and a variant of orf138 coding sequence has lost its ability to induce male sterility [33]. Li et al. [34] detected seven orfH79 haplotypes in accessions of four Oryza species (i.e. Oryza meridionalis, Oryza nivara, Oryza barthii and Oryza rufipogon) among forty-two accessions of five wild rice species collected by the International Rice Research Institute (IRRI). The rice CMS gene, orf352, is also carried by O. rufipogon with several amino acid haplotypes [35]. He et al. [36] has analyzed the sequence including the B-atp6-orf79 like structure and the atp6 in the GenBank database.
Wild rice (O. rufipogon Griff.) is the closest wild relative of Asian cultivated rice (Oryza sativa L.) and has been considered as an ancestral progenitor of the O. sativa [37,38,39,40]. China is an important center of diversity in the extensive distribution of O. rufipogon. The latitudinal range between 18°17′ and 28°05′ N is the most abundant distribution area of O. rufipogon in China, and the resident populations formerly harbored luxuriant genetic diversity [41, 42]. For example, the CMS-HL cytoplasm resource is derived from O. rufipogon with a red awn distributed on Hainan Island, China [22].
The genes orf79, orfH79, and L-orf79 are responsible for CMS-BT [1, 4, 12, 43], CMS-HL [3, 8], and CMS-Lead (Lead type CMS) [2], respectively. Particularly, L-orf79 gene is also detected in the CMS-Liao (Liao type CMS) cytoplasm type, but it is not identified as its sterility gene [44]. L-orf79 gene sequence have no difference in sequence between CMS-Lead and CMS-Liao lines. The orfH79 gene and its variants (hereafter abbreviated as GSV for convenience) are adjacent to and co-transcribed with B-atp6, and it’s a 52 bp downstream connected by an abundant variance 176 bp sequence, which thus constitute the B-atp6-orfH79 region. B-atp6-orfH79 and its variants (hereafter abbreviated as B-atp6-GSV) is transcribed as a single B-atp6-GSV RNA molecule [45,46,47,48]. The sequence structure analysis of B-atp6-GSV would provide an improved understanding the origin of CMS gene, and the molecular mechanism of mtDNA recombination.
The mechanisms of mtDNA recombination provide insight into the causes of its complex structure [49]. In addition to the important application for hybrid seed production, CMS also can be used to research mtDNA transfer [50], the interaction of nuclear DNA and mtDNA, the origin and maintenance of stable gynodioecy in plants [29, 51, 52], and floral organ development [21, 53]. In this study, we surveyed the CMS genetic differentiation among seventeen populations of O. rufipogon across its entire distribution range in China. Gene-specific primers were used to amplify B-atp6-orfH79, orfH79 and atp6 through polymerase chain reaction (PCR), and their sequences were analyzed to address the following aims: (1) to determine if a natural population in China carries the gametophytic CMS gene or variants and, if so, to explore its evolutionary relationships among different haplotypes; (2) to determine the distribution of haplotypes among populations and identify additional cytoplasm resources for rice breeding; (3) to analyze the sequence structure characteristics and uniformity of the chimeric trait of B-atp6-orfH79.
Results
Detection of atp6, B-atp6-orfH79 and orfH79 in Oryza rufipogon
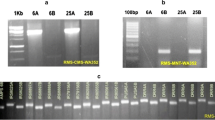
In this study, each individual in all populations can be amplified products using the primers for the atp6 gene (Table 1), indicating the wide presence of atp6 gene in O. rufipogon populations, which was consistent with the previous study that mtDNA is conservative [19]. To verify whether orfH79 and its variants (GSV) were always accompanied by B-atp6, the specific primers for orfH79 and B-atp6-orfH79 were employed. Both primer pairs amplified products for each individual of the HK, GZ, PS, TL, and YJ populations (Table 1), which indicated that the GSV were always accompanied with B-atp6. Conversely, PCR products were not amplified with both primer pairs for the remaining twelve populations, i.e., NHNC, WN, QH, NN, LB, XZ, HZ, GP, YL, FC, BH and DX. The detection of orfH79 gene from the five populations of O. rufipogon in China at a medium frequency about 30%, provided abundant potential CMS sources for rice breeding.
As conserved gene sequence, no differences were found among all the sequences of atp6 in the O. rufipogon populations. Fifteen individuals were randomly selected for sequencing from each population, except for the YJ population with only five individuals. The sequencing results revealed that only one haplotype was identified in all individuals of one population.
Among five populations, three haplotypes (named as H1, H2, and H3, respectively) were detected with the primers for orfH79, and three haplotypes (named as BH1, BH2, and BH3, respectively) were detected with the primers for B-atp6-orfH79. Specifically, The H1, H2, and H3 sequences were identical to the corresponding sequences of BH1, BH2, and BH3 in each population, respectively (Table 2, Table 3 and Fig. 1). For instances, the PS and GZ populations shared H1 and BH1, while the TL and HK populations shared H2 and BH2. Furthermore, H3 and BH3 were only detected in the YJ population.

Collection localities in China, and the distributions of the three mitochondrial haplotypes in seventeen Oryza rufipogon populations
Sequence structure characteristics of B-atp6-GSV, phylogenetic analysis and the uniform chimeric trait
B-atp6-orf79, B-atp6-orfH79, and B-atp6-L-orf79 are previously published haplotypes in O. rufipogon [1,2,3,4]. Among three detected haplotypes in this research, BH2 was identical to B-atp6-orfH79, whereas BH1 and BH3 were novel haplotypes in O. rufipogon populations. B-atp6-orf79, B-atp6-orfH79, B-atp6-L-orf79, BH1, and BH3 constitute the B-atp6-GSV region. Previously, B-atp6-orf79 has been detected in the CMS-Dian 1 and CMS-BT cytoplasm types, and B-atp6-L-orf79 was detected in the CMS-Lead and CMS-Liao types [4, 44]. A total of eighteen haplotypes (i.e. BH1-BH18) were summarized from the GenBank, and their common characteristics and unique variation were demonstrated in Fig. 2. Their accession number in the GenBank of NCBI database, and other information such as population and species have been provided in Additional file 1: Table S1. All were chimeric sequences, containing a 671 bp conserved sequence composed of B-atp6 (619 bp) and 52 bp downstream of the B-atp6 (DS). B-atp6 and DS was identical to the corresponding sequences of atp6 and its corresponding downstream of O. sativa and O. rufipogon [54]. There were no differences in sequence among these eighteen haplotypes, except for BH18 and a single nucleotide polymorphism in 668 position (C turned into G) of BH10. The complex variable sequence (VS) connected with the DS and GSV, is a 176 bp sequence including five insertion or deletion and more than 30 single nucleotide polymorphisms, as shown in Fig. 2.

The sequence structure characteristics of mitochondrion B-atp6-orfH79 and its variants (B-atp6-GSV) (i.e. from BH1 to BH18) of Oryza in sampled population from China and collected from GenBank
Maximum-Likelihood Phylogenies analysis of B-atp6-GSV of eighteen haplotypes (Fig. 3) indicated that three clades are formed with a high support rate. Owing to primer selection, the last 26 bases of GSV were not amplified. Thus these 26 bases were not included in the present and subsequent analysis. BH16 and BH17 formed a clade, while BH1, BH2 and BH13-15 formed another clade. Additionally, the other haplotypes (i.e. BH3-BH12, BH18) formed the third clade (Fig. 3). The new haplotype BH1 was close to the BH13, while another new haplotype BH3 and the existing BH12 clustered together as a sub-clade. BH16 and BH17 formed a clade with no sequential relationship, and both only exist in O. rufipogon distributing in south Asia (only 4 time were detected). Specifically, BH17 was only detected in Thailand, while BH16 was detected in India and Sri Lanka [36]. Additionally, the BH1-2 and BH13-15 formed another clade with a high support rate of each branch, which all were only detected in O. rufipogon. As a new haplotype, BH1 is located on Chinese mainland such as Guangdong Province, while the BH2 haplotype both distribute in Chinese mainland and Hainan Island, including Zhenjiang Town of Guangdong Province and Longhua District of Hainan Province (Fig. 1). Generally, the BH2, located in the south of BH1 in the distribution of populations, also was reported in Thailand. The BH13 was located in China, while both BH14 and BH15 are located in India [36]. However, the WN and QH population close to the BH13 location, did not carry the B-atp6-GSV, indicating its independent occurrence.

Maximum likelihood tree constructed by model HKY85 using eighteen haplotypes of B-atp6-orfH79 from three species of Oryza (the 5’ last 26 bp of the orfH79 was not used in analysis)
The third clade has a support rate of 100% that all the haplotypes from O. sativa gather in these branches, while the support rate is relatively low inside the clade. The BH6 and BH8, located in India (They were detected once, respectively), were only carried by O. rufipogon, and BH4-5 and BH9-10 are grouped into one branch and carried by O. rufipogon, O. sativa or O. bathii, respectively. The BH4 haplotype could be found in all these three Oryza species, while BH5 and BH9 haplotypes were carried by both O. rufipogon and O. sativa. However, the BH10 haplotype only could be found in O. sativa grown in Australia. Except for the BH6, the other haplotypes detected in O. sativa are located in this third-level clade. All the haplotypes carried by Australia O. sativa distribute in this clade. For BH5, the sources of O. sativa are very wide. The O. rufipogon populations distributing in India, Bangladesh and Thailand carried BH4, BH5, BH9 haplotypes, respectively. The BH7 haplotype was found in O. rufipogon populations from India, and the O. sativa populations from Philippines and Africa (Nigeria, Madagascar) [36]. The BH11-12 haplotypes were only detected in China. Apart from one material from Thailan, the BH18 haplotype was only found in the materials from China. As a novel haplotype only found in Yunnan Province, China, the forming reason of BH3 needs to be further studied. Generally, the geographical distribution of haplotypes has certain geographical characteristics. Nonetheless, the B-atp6-GSV does not obey the distribution of general geography. There are certain species boundaries, but not absolutely. For example, all haplotypes from O. sativa are clustered in the third branch, which also could be found in the wild rice.
The populations carrying the B-atp6-GSV appear irregularly, have several different phenotypes (Additional file 2: Fig. S1.). The enough haplotypes of B-atp6-GSV provide a clear structure, including their consistency and variation. We speculated that B-atp6 has the uniform chimeric trait in B-atp6-GSV structure, because of the inverted repeat sequence like GGGCGGGGG……GGGGGCGGG in the B-atp6 structure. Li et al. [34] reported additional seven haplotypes of orfH79, i.e., W11, W15(34), W20, W21, W29, W34, and W46, together with the GSV section in eighteen BH haplotypes of B-atp6-GSV that could be divided into ten GSV haplotypes that belong to three species (i.e. O. rufipogon, O. nivara and O. sativa). Seventeen haplotypes have been found in the GSV section belonging to six species of Oryza (Table 4), due to the completely identical sequences, which demonstrated the inconformity to the classification results of B-atp6-GSV. Concretely, BH4-BH6, BH8-BH10 have the same GSV sequence to W46 and the orf79, while BH2, BH14, BH15 have the same GSV sequence to W42, W45, YtA, and the orfH79. Additionally, BH7 is the same as the L-orf79. All these seventeen GSV haplotypes can be translated into eleven different amino acid sequences as the variation of W34, W46, LR794109 and orf79 codes for the same amino acid sequence (Fig. 4). Haplotype W20 was identical to W15 and W42 except for the variation in the 26 bases. The first 34 bases of GSV, identical to the corresponding sequence of cytochrome oxidase subunit II (COXII), was shared with nuclear DNA, which indicated that all GSV are chimeric sequences.

Deduced amino acid sequences of the seventeen haplotypes VGS (orfH79 and its variants)
Population differentiation
The hierarchical analysis of molecular variance (AMOVA) indicated the occurrence of variation among all populations (FST = 1; all partitions were significant at P < 0.001). According to the VS region of B-atp6-GSV (Fig. 2), the YJ population was formed as group 1, while other populations including the HK, GZ, PS, and TL populations, comprised as group 2. Of the total variation, 33.3% was partitioned among groups and 66.7% among populations within the groups (all partitions were significant at P < 0.001) (Table 5). Furthermore, there was no variation found within each population.
Phylogenetic reconstruction for GSV
In this study, three GSV haplotypes including H1, H2 (orfH79) and H3 were detected in O. rufipogon. In addition, orf79 and L-orf79 are well-known gametophytic CMS genes. For example, orf79 is the sterility gene of CMS-BT and CMS-Dian1 (Dian-1 type CMS), while L-orf79 is the sterility gene of CMS-Lead. Furthermore, W11, W21, W29, W34, W42, and W46 are different variants of orfH79 in the wild rice [34]. As shown in Fig. 5, the maximum likelihood (ML) analysis could not reflect the genetic relationship among six different species according to their comprehensive character. The TL population carried the H2 (orfH79) haplotype, its average kernel set just 4.6%, and the seed setting rate of 73.08% individual was 0 (Additional File 3: Table S2). The PS and GZ population carried the H1 haplotype, and their average kernel were 33.6% and 19.2%, respectively. Corresponding amino acids of variable nucleotide sites for each haplotype of GSV are shown in Fig. 4, and the variable nucleotide sites are shown in Table 4. Overall, single nucleotide polymorphisms of 7 loci bring eleven different amino acid sequences.

Maximum likelihood tree constructed by model HKY85 using seventeen VGS (orfH79 and its variants) of from 6 species of Oryza (the 5’ last 26 bp was not used in analysis)
Discussion
Gametophytic CMS genes and variants may be applied as novel original CMS sources
The BH2 haplotype, detected in the HK and TL populations (red circles in Fig. 1), was identical to B-atp6-orfH79, which is consistent with the previous finding that the CMS-HL source originated from the distant hybridization between wild rice (O. rufipogon from Hainan Province, China) and O. sativa subsp. indica ‘Liantangzao’ [22]. The HK population grown in Hainan Island, China with erect and stout stem, harbors the BH2 haplotype, that do not flower in October. However, despite the identical haplotype, the TL population in Guangdong Province, China could produce thin stolons and flowers in October. These populations can serve as the novel original CMS sources. Until now, L-orf79, orfH79, and orf79 have been considered as gametophytic CMS genes. Each variant haplotype of orfH79 represents a novel CMS type [34], which suggested that expression of the gametophytic CMS genes and the pollen-sterility phenotype resulted from the loss of relevant restorer-of-fertility genes during the distant hybridization. PS, GZ, and YJ populations carrying BH1 and BH3 haplotypes can provide novel original CMS sources for further investigation. A random survey of TL, PS and GZ populations all located in Guangdong province, indicated that the seed setting rate varied significantly (Additional file 3: Table S2, Table S3 and Table S4). For example, the kernel set in TL population was just 4.6%, while the kernel set in PS population reached 33.6%, which should be attributed to the negative impact of environment, climate and temperature on pollen development, or pollen sterility caused by the gametophytic sterility gene.
Geological and ecological factors may contribute to the unique haplotype
Generally, the distribution of haplotypes has certain geographical characteristics. However, the B-atp6-GSV does not completely obey the distribution of general geography. For examples, the first clade distribute in south Asia. However, the haplotypes of the second clade found in China and India (BH1-2 and BH13 from China, but BH14-15 from India), respectively, indicated their certain and limited geographical characteristics. In this study, the HK population is located in the north of Hainan Island, China, whereas the TL population is situated in southern continental China. Although these two populations are separated by the South China Sea and Leizhou Peninsula (Fig. 1), both populations harbored the BH2 haplotype. Despite the close geographic proximity among the GZ, TL and PS populations with a mutual linear distance below 10.0 km (Table 6 and Fig. 1), the GZ and PS populations shared the BH1 haplotype, whereas the TL population harbored BH2 haplotype. Additionally, the BH2 was also detected in the north of the PS population. The above phenomenons and results indicated the genetic recombination among various populations independently. The high support rate of the ML tree of the B-atp6-GVS illustrated that BH1and BH2 forms different taxon with high support, respectively. Conversely, the BH3 haplotype was only detected in the YJ population of wild rice, that is located in Yuanjiang of Yunnan Province, China. All of these populations grow in transitional areas between subtropical and tropical climates [55]. We inferred that these unique haplotypes were produced because of the geological and ecological niches of the transitional area between the tropical and subtropical climatic zones. A unique haplotype coincident with geological and ecological factors has been reported previously in yews [56]. Unfortunately, the HK population is extinct with no individuals of O. rufipogon as a result of real estate development and duck farming, when we returned to the sampling site in January 2016, the second year of our sampling. Protection of the TL population, which also harbors B-atp6-orfH79, is a matter of urgency. The origin of the unique BH3 haplotype in Yuanjiang of Yunnan Province, deserves further study.
The uniform chimeric trait
Eighteen haplotypes of B-atp6-GSV have been identified at present (Fig. 2). Particularly, BH1 and BH3 were first distinguished in this present study, whereas B-atp6-orf79, B-atp6-orfH79, and B-atp6-L-orf79 have been identified previously and correspond to the CMS-BT, CMS-HL, and CMS-Lead genes, respectively [43, 48]. The original CMS-BT source arose through interspecific hybridization between O. sativa subsp. indica ‘Chinsurash Boro II’ (origin in India) and O. sativa subsp. japonica ‘Taizhung 65’ [25]. The original CMS-Dian 1 source was bred from sterile plants of ‘Taibei 8’ (a japonica rice grown at low altitude) planted in Baoshan, Yunnan Province in 1965. The sterile individual was speculated to be a result of the spontaneous hybridization between ‘Taibei 8’ and non-glutinous rice (O. sativa L.) grown at high altitude [15]. Orf79 is also considered to be associated with CMS-Dian 1 [48]. The original CMS-HL source arose through the distant hybridization between wild rice (grown at Lingshui County, Hainan Island, China) and O. sativa subsp. indica ‘Liantangzao’ [22]. The original CMS-Lead source was identified by Watanabe (1971) [57], who hybridized O. sativa from Myanmar, ‘Lead Rice’, and the japonica cultivar ‘Fujisaka 5’ [46]. The original source of CMS-Liao line has been developed using the cytoplasm of ‘IR24’ [58].
Among the aforementioned plant material, ‘Chinsurash Boro II’ (planted in India), ‘Taibei 8’ (planted in Taiwan, China), ‘Lead Rice’ (planted in Myanmar), ‘IR24’, and the YJ, HK, TL, PS, and GZ populations all harbor B-atp6-GSV with wild distribution and geographical separation. Based on the different haplotypes of B-atp6-GSV and their distribution and the phenotypes of O. rufipogon populations, and the ML analysis, we speculated that the different haplotypes of B-atp6-GSV did not show a single, common origin and the chimerism of B-atp6-GSV occurs independently. The sequence structure of B-atp6-GSV includes a conserved sequence 5′-GGGCGGGGG……GGGGGCGGGACAAA-3′ (671 bp), of which GGGCGGGG……TGAGTAA (B-atp6, 610 bp) identical to the corresponding sequence of rice atp6, and TTTCATAA……GGGGGCGGGA (52 bp) that is a downstream of the rice atp6 (Fig. 2) [54]. The different haplotypes of B-atp6-GSV occurred independently, indicating that 5′-GGGCGGGGG-3′ and 5′-GGGGGGCGGGA-3′ are the sites prone to chimerism, which was closely associated with the origin of the gametophytic CMS gene. The frequent insertion or deletion of VS segments is the same as the recombination site of nuclear DNA. The high support rate of B-atp6-GSV (Fig. 3) and the low support rate in phylogeny tree of GSV (Fig. 5) supported this view. The high support rate of the B-atp6-GSV, may be due to the abundant variation sites and long enough sequences, so that the phylogenetic tree can be established more accurately. Due to lack of rich enough variation sites in GSV section, the phylogenetic tree could not reflect the true genetic relationship of different species for the low support rate.
Until now, based on the high homologous sequences in GenBank of NCBI database, eighteen various haplotypes of B-atp6-GVS structure including two novel haplotypes (i.e. BH1 and BH3) detected in this study, have been summarized for the ML analysis [36]. Among them, the populations carrying the BH1, BH2 and BH3 formed different clade with a high support rate of 100%, which means their independent occurrence, and doesn't happen randomly. Finding the B-atp6-GVS unified chimeric site is very important to explore the molecular mechanism of gametophytic sterility gene production, and understand the mechanism of chimerism, so as to advance the knowledge about the genetic and evolutionary potential principle of chimerism. Only with enough haplotypes, we can find relatively consistent chimeric sites. The haplotypes in this paper is the most haplotype of known chimeric structure. By exploring the chimeric mechanism of sterile genes, we may be able to understand the reasons for the frequent recombination of mtDNA, which might promote the creation of original CMS and understand the mtDNA well.
All these haplotypes show the uniform chimeric trait. GGGCGGGGG……GGGGGCGGG is a similar 123,456,789……987,654,321 unified transposable substructure. The abundant variation sites in the VS region should be attributed to the recombination repair mechanism, which contributed to gain insight into the molecular mechanism of mtDNA chimera formation. Terminal inverted repeats characteristically flank each end of DNA transposons [59, 60]. Transposons are an important source of genetic novelty [61].
Sequences shared by mitochondrial and nuclear genomes
B-atp6 is identical to the corresponding sequence of the rice mitochondrial gene atp6. The nuclear gene atp6 is located on chromosome 1 of the ‘Nipponbare’ nuclear genome (AP014957.1) as a highly conserved region [62, 63]. The gene atp6 also have be found in chromosome 6 of Minghui 63 nuclear genome, chromosome 6 of Shuhui498 for two times, chromosome 1 of Oryza sativa Japonica and Zhenshan 97, and chromosome 11, respectively [64,65,66]. This study confirmed that the VS sequence (176 bp) contains about five insertion or deletion, and more than 30 single nucleotide polymorphism. The first 85 bases of COXII are conserved among all different species, which is shared by the mitochondrial and nuclear genomes. The first 34 bases of GSV are identical to the corresponding sequence of COXII. COXII introns are reported to undergo frequent horizontal transfer [67]. The sequences shared by the mitochondrial and nuclear genomes are responsible for reproductive barriers in rice [68]. Additional research is needed into whether the sequences shared by the mitochondrial and nuclear genomes are associated with the origin of CMS genes and chimeric events.
Evolution of GSV
The different components of GSV differ in their effect. Pollen of CMS-HL aborts at the binucleate stage, while CMS-BT pollen aborts at the trinucleate stage. Furthermore, pollen abortion occurs at a later stage in the CMS-Lead line than in the CMS-BT line. All three CMS lines show gametophytic sterility [43, 44]. The basal nodes of the phylogenetic tree derived from GSV sequence data cannot reflect the development and evolution of the pollen phenotype. H2 in the top of the phylogenetic tree of GSV, and the H1 is closer to the base. In the process of creating the original CMS source, the parent material initially seed setting normally, the seed setting rate decreased gradually with the breeding crossing, finally the CMS line was bred. It seem as the relationship between H1 and H2 was reflected on the phylogenetic tree of GSV. However, under normal circumstances, we think plant do not allow itself to evolve to extinct. These contradictions reflect the complexity of the evolution of plant mtDNA. The YJ population harboring H3 haplotype shows normal seed set through natural pollination. The GSV haplotype W11 is carried by the Oryza meridionalis grown in Australia, the GSV haplotype W21 is carried by the Oryza barthii grown in Nigeria, and W34 is carried by Oryza nivara-Ura Wee grown in Sri-Lawka, while the GSV haplotype W15 and W29 are carried by the Oryza nivara grown in India, and W42 and W46 are carried by O. rufipogon grown in Laos and Comodia, respectively [34]. All these materials belong to Oryza AA genome. Despite the abundant variation sites in GSV, the close relationship between O. niwara and O. rufipogon can not be reflected by the ML tree [69] with a relatively low support rate, which should be attributed to the short sequence in GSV with only about 200 bp. On the other hand, at present, their unclear sources also restricted the origin research of gametophytic CMS gene in wild rice.
Conclusions
In this work, the B-atp6-orfH79, orfH79 and atp6 in 427 individuals of seventeen O. rufipogon populations in China were amplified using specific primers through PCR, demonstrating that the gametophytic CMS genes carried by O. rufipogon are distributed in transitional geological and ecological niches. Two novel detected haplotypes (i.e. BH1 and BH3) of B-atp6-orfH79 provided original CMS sources for rice breeding. The sequence characteristics of B-atp6-orfH79 and ML analysis indicated the chimerise occur independently with consistent chimeric sites, which might help to explore the origin of rice gametophytic CMS genes in O. rufipogon. Uniform chimeric traits in the rice atp6 gene accompany the gametophytic CMS genes. GSV is always accompanied by B-atp6 in the natural population of O. rufipogon.
Materials and methods
Plant materials collected in China
A total of 427 individuals with seventeen populations, were identified as O. rufipogon by Xuemei Zhang with the assistance of Yating Liu and relevant experts according to its morphological identification, which were collected in from Dongxiang of Jiangxi Province to Sanya of Hainan Province since July 2013 to November 2015 (Fig. 1 and Table 6), representing the geographical distribution of this species in China. Their growing environment and plant appearance of seventeen Oryza rufipogon populations were presented in Additional file 2: Figure S1. Samples were randomly collected from individuals separated by at least 5 m to prevent collection of multiple samples from a single genet, except for the NN and YJ populations that the young and healthy leaves of all surviving individuals were sampled, because of the extremely endangered. The collected young and healthy leaves were immediately desiccated in silica gel for DNA extraction.
DNA extraction
Total genomic DNA was extracted from the desiccated leaf tissue using the cetyltrimethylammonium bromide extraction method established by Doyle & Doyle (1987) [70], and an ammonium acetate wash for additional purification [71]. The DNA was dissolved in 1 × Tris–EDTA buffer (10 mmol/L Tris–HCl, 1 mmol/L EDTA, pH 8.0) to a final concentration of 20–40 ng/µL.
PCR amplification and sequencing
Specific primers for GSV and B-atp6-GSV [8] were used to detect the origin of the gametophytic sterility gene and variation in the sequence structure of B-atp6-GSV. Primers for atp6 amplification were tested on each of the 427 individuals as a positive control for the gametophytic CMS gene. The PCR mixture (total volume 25 µL) contained 2.5 µL of 10 × PCR buffer, 2.5 µL MgCl2 (25 mM), 2.0 µL dNTPs mixture (2.5 mM), 0.3 µL each primer (10 µM), 0.2 µL Taq polymerase (5 U/µL) (Sangon Biotech, Shanghai, China), 1 µL template DNA (~ 20–40 ng genomic DNA), and 16.2 µL distilled deionized water. The PCR amplifications were performed on a GeneAmp PCR System 9700 thermal cycler (Perkin Elmer, Foster, CA, USA) with the following protocol: initial denaturation at 95 °C for 4 min, followed by 30 cycles of 95 °C for 1 min, 50–56 °C for 1.5 min, 72 °C for 2 min, and final elongation at 72 °C for 5 min. Amplification products were visualized by 1% agarose gel electrophoresis. If an entire population yielded amplified products, 15 individuals per population were randomly selected for sequencing to evaluate their sequence variations. If a population comprised less than 15 individuals, each individual of the population was sequenced. The PCR products were analyzed and sequenced using an ABI 3700 automated sequencer (Applied Biosystems, Waltham, MA, USA) with the assistance from Sangon Biotech Co., Ltd (Shanghai, China). Contiguous nucleotide sequences were edited using SeqMan in the LaserGene package (DNA Star, Inc., Madison, WI, USA). Consensus sequences were aligned using ClustalX [72] and then adjusted manually.
Data analysis
The amount of variation among populations within regions and within populations was calculated by means of a hierarchical AMOVA framework [73] using Arlequin 3.0 [74]. Significance was tested using a non-parametric permutation procedure with 1000 permutations. The ML tree was constructed based on evolution distance from the multiple alignment of GSV and B-atp6-GSV sequences by PhyML 3.0 software using the maximum likelihood method [75]. The geographical distribution of the mitochondrial haplotypes was mapped by using ArcMap 9.1 (ESRI, Redlands, CA, USA). The BLASTX tool translated the nucleotide sequences into amino acid sequences, and the sequence identities were calculated by the Global Alignment. Additionally, the nucleotide BLAST was used to search for homologous sequences with default parameters in GenBank of NCBI database (https://www.ncbi.nlm.nih.gov).
Availability of data and materials
The collected plant materials have been deposited in specimen room of The State Key Laboratory of Conservation and Utilization of Bio-resources in Yunnan for the public exhibitions and preservation. All data generated or analyzed during this study are included in this published article. The relevant sequences are available in the GenBank of NCBI database with the accession numbers from KY856719 to KY856721.
Abbreviations
- CMS:
-
Cytoplasmic male sterility
- mtDNA:
-
Mitochondrial DNA
- CMS-WA:
-
Wild Abortive type CMS
- CMS-BT:
-
Boro II type CMS
- CMS-HL:
-
HongLian type CMS
- PCR:
-
Polymerase chain reaction
- IRRI:
-
International Rice Research Institute
- GSV:
-
orfH79 And its variants
- B-atp6-GSV:
-
B-atp6-orfH79 and its variants
- DS:
-
Downstream of the B-atp6
- VS:
-
Variable sequence
- CMS-Dian1:
-
Dian-1 type CMS
- CMS-Lead:
-
Lead type CMS
- CMS-Liao:
-
Liao type CMS
- COXII:
-
Cytochrome oxidase subunit II
- EDTA:
-
Ethylene diamine tetraacetic acid
- BLAST:
-
Basic Local Alignment Search Tool
- AMOVA:
-
Analysis of molecular variation
- ML:
-
Maximum likelihood
- NCBI:
-
National Center for Biotechnology Information
References
Iwabuchi M, Kyozuka J, Shimamoto K. Processing followed by complete editing of an altered mitochondrial apt6 RNA restores fertility of cytoplasmic male sterile rice. EMBOJ. 1993;12:1437–46.
Kazama T, Itabashi E, Fujii S, Nakamura T, Toriyama K. Mitochondrial ORF79 levels determine pollen abortion in cytoplasmic male sterile rice. Plant J. 2016;85:707–16.
Mireau H. Turning an essential respiratory gene into a cytoplasmic male-sterility factor. Mol Plant. 2022;15(6):931–3.
Wang ZH, Zou Y, Li X, Zhang Q, Chen L, Wu H, et al. Cytoplasmic male sterility of rice with Boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell. 2006;18:676–87.
Kazama T, Okuno M, Watari Y, Yanase S, Koizuka C, Tsuruta Y, et al. Curing cytoplasmic male sterility via TALEN-mediated mitochondrial genome editing. Nat Plants. 2019;5:722–30.
Young EG, Hanson MR. A fused mitochondrial gene associated with cytoplasmic male sterility is developmentally regulated. Cell. 1987;50(3):41–9.
Das S, Sen S, Chakraborty A, Chakraborti P, Maiti MK, Basu A, Basu D, Sen SK. An unedited 1.1 kb mitochondrial orfB gene transcript in the wild abortive cytoplasmic male sterility (CMS-WA) system of Oryza sativa L. subsp. Indica. BMC Plant Biology. 2010;10:39–57.
Hu J, Wang K, Huang WC, Liu G, Gao Y, Wang JM, et al. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell. 2012;24:109–22.
Luo DG, Xu H, Liu ZL, Guo JX, Li HY, Chen LT, et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat Genet. 2013;45:573–7.
Fujii S, Yamada M, Fujita M, Itabashi E, Hamada K, Yano K, et al. Cytoplasmic-nuclear genomic barriers in rice pollen development revealed by comparison of global gene expression profiles among five independent cytoplasmic male sterile lines. Plant Cell Physiol. 2010;51(4):610–20.
Bentolila S, Stefanov S. A reevaluation of rice mitochondrial evolution based on the complete sequence of male fertile and male sterile mitochondrial genomes. Plant Physiol Preview. 2012;158(2):996–1017.
Kazama T, Toriyama K. Whole mitochondrial genome sequencing and re-examination of a cytoplasmic male sterility-associated gene in Boro-Taichung-type cytoplasmic male sterile rice. PLoS ONE. 2016;11(7): e0159379.
Mogensen HL. The how and whys of cytoplasmic inheritance in seed plants. Am J Bot. 1996;83(3):383–404.
Mackenzie S, McIntosh L. Higher plant mitochondria. Plant Cell. 1999;11:571–85.
Li ZY. Dian type hybrid rice memoir, Kunming. China: Yunnan people press; 1999.
Albert B, Godell B, Gouyon PH. Evolution of the plant mitochondrial genome, dynamics of duplication and deletion of sequences. J Mol Evol. 1998;46:155–8.
Woloszynska M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes-though this be madness, yet there’s method in’t. J Exp Bot. 2010;61:657–71.
Arrieta-Montiel MP, Mackenzie SA. Plant mitochondrial genomes and recombination. Plant Mitochon. 2011;1:65–82.
Chen LT, Liu YG. Male sterility and fertility restoration in crops. Annu Rev Plant Biol. 2014;65:579–606.
Wynn EL, Christensen AC. Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3-Genes Genomes Genet. 2019;9:549–59.
Gurdon C, Svab Z, Feng Y, Kumar D, Maliga P. Cell-to-cell movement of mitochondria in plants. Proc Natl Acad Sci USA. 2016;113(12):3395–400.
Zhu YG. Rice male sterility biology. Wuhan: China. Wuhan University Press; 2000.
Ogura H. Studies on the new male sterility in Japanese radish, with special reference to the utilization of this sterility towards the practical raising of hybrid seeds. Mem Fac Agric Kagoshima Univ. 1968;6(2):39–78.
Shinjyo. Cytoplasmic genetic male sterility in cultivated rice, Oryza sativa L. II. the inheritance of male sterility. Jap J Genet. 1969;44:149–56.
ICRISAT (International Crops Research Institute for the Semi-Arid Tropics), 1982. Sorghum in the Eighties, Proceedings of the International Symposium on Sorghum, 2–7 Nov 1981, Patancheru. pp 364–378. A.P. India, Patancheru, A.P., India, ICRISAT
Forsberg J, Dixelius C, Lagercrantz U, Glimelius K. UV dose-dependent DNA elimination in asymmetric hybrids between Brassica napus and Arabidopsis thaliana. Plant Sci. 1998;131:65–76.
CAAS & HAAS (Chinese Academy of Agricultural Sciences and Hunan academy of agricultural sciences). Development of hybrid rice in China. Beijing: Agriculture Press; 1991. p:20–30.
Dufaÿ M, Touzet PS, Maurice S, Cuguen J. Modelling the maintenance of male-fertile cytoplasm in a gynodioecious population. Heredity. 2007;99:349–56.
Serieys H, Christov, M. 2005. Progress Report 1999–2004, FAO Working Group, “Identification, Study and Utilization in Breeding Programs of New CMS Sources”, X Consultation Meeting. p 80. Novi Sad, Serbia and Montenegro.
Sandhu A, Abdelnoor RV, Mackenzie SA. Transgenic induction of mitochondrial rearrangements for cytoplasmic male sterility in crop plants. Proc Natl Acad Sci. 2007;104(6):1766–70.
Rieseberg LH, Fossen CV, Adrias D, Carter RL. Cytoplasmic male sterility in sunflower, origin, inheritance, and frequency in natural populations. J Hered. 1994;85:233–8.
Terachi T, Yamaguhi K, Yamagishi H. Sequence analysis on the mitochondrial orfB locus in normal and Ogura male-sterile cytoplasms from wild and cultivated radish. Curr Genet. 2001;40:276–81.
Giancola S, Rao Y, Chaillou S, Hiard S, Martin-Canadell A, Pelletier G, et al. Cytoplasmic suppression of Ogura cytoplasmic male sterility in European natural populations of Raphanus raphanistrum. Theor Appl Genet. 2007;114:1333–43.
Li SQ, Tan YP, Wang K, Wan CX, Zhu YG. Gametophytically alloplasmic CMS line of rice (Oryza sativa L.) with variant orfH79 haplotype corresponds to specific fertility restorer. Theor Appl Genet. 2008;117:1389–97.
Tang H, Zheng XM, Li CL, Xie XR, Chen YL, Chen LT, et al. Multi-step formation, evolution, and functionalization of new cytoplasmic male sterility genes in the plant mitochondrial genomes. Cell Res. 2017;27:130–46.
He W, Chen C, Adedze YMN, Dong X, Xi K, et al. Multicentric origin and diversification of atp6-orf79-like structures reveal mitochondrial gene flows in Oryza rufipogon and Oryza sativa. Evol Appl. 2020;13:2284–99.
Zhu QH, Zheng XM, Luo JG, Gaut BS, Ge S. Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol. 2007;24(3):875–88.
Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Q, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501.
Vaughan DA, Morishima H. Biosystematics of the genus Oryza. In: Smith CW, editor. Rice: origin, history, technology, and production. Hoboken, NJ: John Wiley & Sons, Inc.; 2003. p. 27–65.
Khush GS. Origin, dispersal, cultivation and variation of rice. Plant Mol Biol. 1997;35:25–34.
NEGWR (National Exploration Group of Wild Rices). An investigation of genetic resources of wild rices in China. Acta Agric Sinica. 1984;6:3–10.
Gao LZ, Ge S, Hong DY. High levels of genetic differentiation of Oryza officinalis Wall et Watt from China. J Hered. 2001;92:511–6.
Gualberto JM, Newton KJ. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu Rev Plant Biol. 2017;68:225–52.
Luan J, Liu TR, Luo WQ, Liu W, Peng MQ, Li WJ, et al. Mitochondrial DNA genetic polymorphism in thirteen rice cytoplasmic male sterile lines. Plant Cell Rep. 2013;32(4):545–54.
Peng XJ, Wang K, Hu CF, Zhu YL, Wang T, Yang J, et al. The mitochondrial gene orfH79 plays a critical role in impairing both male gametophyte development and root growth in CMS-Honglian rice. BMC Plant Biol. 2010;10:125.
Itabashi E, Kazama T, Toriyama K. Characterization of cytoplasmic male sterility of rice with lead rice cytoplasm in comparison with that with Chinsurah Boro II cytoplasm. Plant Cell Rep. 2009;28:233–9.
Fujii S, Kazama T, Yamada M, Toriyama K. Discovery of global genomic re-organization based on comparison of two newly sequenced rice mitochondrial genomes with cytoplasmic male sterility-related genes. BMC Genomics. 2010;11:209.
Zhu GQ, Tan XL, Zhao Y, Zi QY, Zheng YZ, Yan CQ, et al. Relationship between CMS-specific mitochondrial structures and pollen abortive phenotype in rice CMS lines. Euphytica. 2015;206(1):149–58.
Alverson AJ, Rice DW, Dickinson S, Barry K, Palmer JD. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber (Cucumis sativus). Plant Cell. 2011;23(7):2499–513.
Bergthorsson U, Adams KL, Thomason B, Palmer JD. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature. 2003;421(10):197–201.
Chase CD. Cytoplasmic male sterility, a window to the world of plant mitochondrial–nuclear interactions. Trends Genet. 2007;23(2):81–90.
Jin Z, Seo J, Kim B, Lee SY, Koh HJ. Identification of a candidate gene for the novel cytoplasmic male sterility derived from intersubspecific crosses in rice (Oryza sativa L.). Genes. 2021;12:590.
Liu B, Ou CG, Chen SM, Cao QW, Zhao ZW, Miao ZG, et al. Differentially expressed genes between Carrot petaloid cytoplasmic male sterile and maintainer during floral development. Sci Rep. 2019;9:17384.
Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, et al. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome, frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Genet Genomics. 2002;268:434–45.
Wu ZY, Wang HS. Chinese physical geography—phytogeography (I). Beijing: Science Press; 1983.
Liu J, Michael M, Jim P, Gao LM, Ram CP, Li DZ. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phyto. 2013;199:1093–108.
Yang MX, Jiang H, Tian Z, Liu XQ, Tan YP, Wang CT. Function of mitochondrial gene orf290 in rice. J Huazhong Agric Univ: Nature Science ED 2018;37(6):1–6 (In Chinese).
Deng H. Three lines hybrid japonica rice breeding. In: Deng H, Hua Z, Yang F, editors. Japonica hybrid rice in China. Beijing, China: China Agriculture Press; 2008. p. 243–71.
Mao L, Wood TC, Yu Y, Budiman MA, Tomkins J, Woo S. Rice transposable elements: a survey of 73,000 sequence-tagged-connectors (STCs). Genome Res. 2000;10:982–90.
Feschotte C, Jiang N, Wessler SR. Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002;3:329–41.
Lipatov M, Lenkov K, Petrov DA, Bergman CM. Paucity of chimeric gene-transposable element transcripts in the Drosophila melanogaster genome. BMC Biol. 2005;3:24.
Feng Q, Zhang Y, Hao P, Wang S, Fu G, Huang Y, et al. Sequence and analysis of rice chromosome 4. Nature. 2002;420:316–20.
KawaharaY, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013; 6 (1):4.
Sasaki T, Matsumoto T, Yamamoto K, Sakata K, Baba T, Katayose Y, et al. The genome sequence and structure of rice chromosome 1. Nature. 2002;420:312–6.
Zhang J, Chen LL, Xing F, Kudrna DA, Yao W, Copetti D, et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc Natl Acad Sci. 2016;113:E5163–71.
Du H, Yu Y, Ma Y, Gao Q, Cao Y, Chen Z, et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun. 2017;8:15324. https://doi.org/10.1038/ncomms15324.
Palmer JD, Adams KL, Cho Y, Parkinson CL, Qiu YL, Song K. Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc Natl Acad Sci USA. 2000;97(13):6960–6.
Yamagata Y, Yamamoto E, Aya K, Win KT, Doi K, et al. Mitochondrial gene in the nuclear genome induces reproductive barrier in rice. Proc Natl Acad Sci USA. 2010;107:1494–9.
Lu BR, Ge S, Sang T, Chen JK, Hong DY. The currenttaxonomy and perplexity of the genus Oryza (Poaceae). Acta Phytotax Sin. 2001;39:373–88.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytocheml Bull. 1987;19:11–5.
Weising K, Nybom H, Wolff K, Meyer W. DNA Finger printing in Plants and Fungi. Boca Raton, Florida USA: CRC Press; 1995.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The clustal x windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–82.
Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distance among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Acknowledgements
We thank Mr. Jian Rong and Pro. Yinhe Zhao for their assistance in this study.
Funding
This research was supported by the National Natural Science Foundation of China (31200908, 8176140709 & 32260681). The funder had no role in the design of the study, in data collection, analysis or interpretation, or in writing the manuscript, but provide the financial.
Author information
Authors and Affiliations
Contributions
Xuemei Zhang conceived and designed the study, analyzed data, and wrote the main manuscript; Shuying Chen and Zixian Zhao performed the experiments; Cunqiang Ma assisted in the data analysis and the paper writing. Yating Liu undertook formal identification of wild rice (Oryza rufipogon Griff.), revised the manuscript, developed the concept and directed the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethics approval and consent to participate are not applicable. Plant samples used in the study were not collected from national park or natural reserve, and fully comply with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. 18 Haplotype of B-atp6-orfH79,and their accession number and relevant population information.
Additional file 2:
Fig. S1. Growing environment and plant appearance of 17 Oryza rufipogonpopulations including BH, DX, FC, GP, GZ, HK, HZ, LB, NHNC, NN, PS, TL, WN, XZ, YJ, YLand QH, that distribute in from northeastern of Jiangxi Province to southeastern of HainanProvince, China.
Additional file 3:
TableS2. Seed setting rate of each individuals collected from TL population located inGuangdong province. Table S3. Seed setting rateof each individuals collected from PS population also located in Guangdongprovince. Table S4. Seed setting rate of sampled individuals collectedfrom GZ population in Guangdong province.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, X., Chen, S., Zhao, Z. et al. Investigation of B-atp6-orfH79 distributing in Chinese populations of Oryza rufipogon and analysis of its chimeric structure. BMC Plant Biol 23, 81 (2023). https://doi.org/10.1186/s12870-023-04082-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04082-5