
Abstract
Background
Plant height is a key factor in the determination of rice yield since excessive height can easily cause lodging and reduce yield. Therefore, the identification and analysis of plant height-related genes to elucidate their physiological, biochemical, and molecular mechanisms have significant implications for rice breeding and production.
Results
High-throughput quantitative trait locus (QTL) sequencing analysis of a 638-individual F2:3 mapping population resulted in the identification of a novel height-related QTL (qPH9), which was mapped to a 2.02-Mb region of Chromosome 9. Local QTL mapping, which was conducted using 13 single nucleotide polymorphism (SNP)-based Kompetitive allele-specific PCR (KASP) markers for the qPH9 region, and traditional linkage analysis, facilitated the localization of qPH9 to a 126-kb region that contained 15 genes. Subsequent haplotype and sequence analyses indicated that OsPH9 was the most probable candidate gene for plant height at this locus, and functional analysis of osph9 CRISPR/Cas9-generated OsPH9 knockout mutants supported this conclusion.
Conclusion
OsPH9 was identified as a novel regulatory gene associated with plant height in rice, along with a height-reducing allele in ‘Dongfu-114’ rice, thereby representing an important molecular target for rice improvement. The findings of the present study are expected to spur the investigation of genetic mechanisms underlying rice plant height and further the improvement of rice plant height through marker-assisted selection.
Similar content being viewed by others


Introduction
Plant height is a key factor in determining rice yield since excessive height can easily cause lodging and reduce yield. As such, the breeding and large-scale promotion of semi-dwarf rice varieties, which has been conducted since the 1950s, has increased rice yield by 20–30% and has, accordingly, been hailed as the "green revolution" in rice production. The first major breakthrough was attributed to the successful application of a semi-dwarf gene (Peng et al. 1999; Hedden 2003).In the 1970s and 1980s, three-line rice breeding aimed at promoting heterosis resulting in another leap in rice yield and progression towards ensuring China’s food security. Nowadays, semi-dwarfing materials are also used in the successful development of hybrid rice. However, in the 30 years succeeding the two major leaps in dwarf and hybrid rice breeding, the yield of rice in China has remained more or less stagnant.
Super rice breeding with ideal plant type as the model represents an important strategy for promoting future improvements in China’s rice yield. The main strategy of such breeding efforts is to combine ideal plant type with heterosis (Chen et al. 2001). Plant type improvement is the main aim of rice breeding and is highly dependent on plant height. However, the semi-dwarf varieties that are currently used in production are all associated with the recessive dwarf gene Ossd1, and the wide application of this single gene poses a potential risk of genetic diversity loss. At the same time, rice varieties that carry the semi-dwarf gene sd1 also exhibit poor drought tolerance and low photosynthetic effect, which has become a bottleneck in the development of new rice varieties (Sha et al. 2021). Therefore, the identification and analysis of plant height-related genes, to elucidate their physiological, biochemical, and molecular mechanisms, have significant implications for rice breeding and production.
Rice plant height is a quantitative trait controlled by multiple genes, that are associated with a variety of physiological traits and processes. In fact, at least 70 dwarf mutants of rice have been discovered, and the underlying mechanisms have reportedly been associated with the phytohormone signaling and the biosynthesis of gibberellic acid (GA), abscisic acid (ABA), and various brassinosteroids (Spielmeyer et al. 2002; Sakamoto et al. 2004; Hubbard et al. 2010; Sun 2010; Song et al. 2012; Shen et al. 2014; Tong et al. 2014; Liu et al. 2015; Wu et al. 2020; Lin et al. 2020); as well as transcriptional regulation, which plays an important regulatory role in rice growth and development (Tan et al. 2008; Yaish et al. 2010; Wu et al. 2020; Wei et al. 2021). The gene IPA1, for example, which encodes a transcription factor that contains the SBP-box domain, has been reported to regulate multiple growth and developmental processes, including the formation of an ideal plant type, and loss-of-function mutants characterized by desirable agronomic traits; such as fewer ineffective tillers, strong stalks, lodging resistance, large panicles with more grains, and high yield (Zhang et al. 2017; Liu et al. 2019; Wang et al. 2020). Meanwhile, Wei et al. (2021) reported the localization and cloning of the plant height-related WRKY-family transcription factor WRKY21. Thus, mining for novel plant height genes is likely an effective strategy for further constructing an ideal plant type and increasing yield.
One effective method for the identification of novel height-related genes is the use ofQTL linkage analysis. Traditional QTL mapping usually involves the genotyping of numerous individuals from a population using molecular markers distributed across the whole genome (Zou et al. 2016). However, to ensure sufficient statistical power, this strategy requires the genotype and phenotype analysis of numerous offsprings, which is both time-consuming and labor-intensive. In contrast, bulked segregant analysis (BSA) only requires the genotyping of individuals with extreme phenotypes (Giovannoni et al. 1991; Michelmore et al. 1991). Initially, BSA was widely used in QTL identification and gene mining related to specific traits; such as disease resistance, color, and fertility (Zhang et al. 1994, 1996; Monna et al. 1995). However, the recent and rapid development of next-generation sequencing (NGS) technology has enabled the use of BSA, along with whole-genome sequencing (i.e., BSA-seq), to efficiently identify QTLs and trait-related genes (Abe et al. 2012; Kadambari et al. 2018). In comparison to traditional QTL mapping, BSA-seq ensures improved work efficiency and sufficient statistical power and has been successfully applied to a variety of plant taxa; including arabidopsis, rice, maize, and pepper (Ramirez-Gonzalez et al. 2015; Huang et al. 2017; Zegeye et al. 2018). However, because the method yields less-than-ideal confidence interval resolution, researchers have had to combine BSA-seq with fine mapping to identify potential candidate genes for specific QTLs (Wambugu et al. 2018). For instance, the QTL qRSL7 (Lei et al. 2020) was mapped from 4.17 Mb to 222 kb by BSA-seq and classical QTL mapping to allow the identification of final candidate genes. In addition, the gene ZmVEN1, which is associated with maize grain texture, was detected using BSA-seq and RNA-seq (Wen et al. 2019). Thus, the integration of BSA-seq and fine mapping are necessary for locating major QTLs and mining target genes.
Accordingly, the aim of the present study was to implement BSA-seq and fine mapping for the identification of height-related genes in rice. An F2:3 population derived from a cross of tall and dwarf varieties (Longyang11 and Dongfu114, respectively) was subjected to BSA-seq analysis and fine-mapping strategy, along with haplotype and sequence analysis, to identify potential candidate genes. Further, CRISPR/Cas9 gene editing was used to develop a knockout mutant to assess the function of the putative height-related genes.
Materials and Methods
Plant Materials and Height Evaluation
The japonica varieties ‘Dongfu 114’ (‘DF114’) and ‘Longyang 11’ (‘LY11’) were obtained from Northeast Agriculture University (Harbin, China) and used as female and male parents, respectively, to generate an F2:3 population of 638 individuals. In the spring of 2019, ‘DF114’ (n = 48), ‘LY11’ (n = 48), and F2:3 (n = 638) individuals were planted in four rows under natural conditions in paddy fields at Acheng Experimental Station (Heilongjiang Province, China), and 5 plants from the center of each plot were selected for plant height evaluation.
Construction of Segregating Pools
All flag leaves of the 638 F2:3 individuals were collected separately for total genomic DNA extraction, which was performed using the Cetyltrimethylammonium bromide (CTAB) method (Murray et al. 1980), with minor modifications, and the isolated DNA was quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). After quantification, a Qubit 2.0 fluorimeter (Life Technologies, Carlsbad, CA, USA) was used to pool equimolar amounts of genomic DNA from the 30 shortest and 30 tallest individuals (L-pool and H-pool, respectively).
Whole-Genome Re-Sequencing
Total genomic DNA was extracted from the bulked pools, and at least 3 µg of genomic DNA was used to construct paired-end libraries, with an insert size of 500 bp, using the Illumina paired-end DNA sample prep kit (San Diego, CA, USA). The resulting libraries were sequenced by Genedenovo (Guangzhou, China) using the HiSeq X10 NGS platform (Illumina). To achieve quality trimming, thereby ensuring high-confidence variant calling, raw reads were filtered by removing reads with ≥ 10% unidentified nucleotides, with > 50% bases with shared quality scores of ≤ 20, or aligned to the barcode adapter. To identify SNPs and indels, filtered reads were aligned to the ‘Nipponbare’ reference genome sequence (Matsumoto et al. 2005) using the Burrows-Wheeler Aligner (v.0.7.16a-r1181; Kumar et al. 2019), using ‘mem -M’ setting, where -M is an option used to mark shorter split-alignment hits as secondary alignments. Variant calling was performed using the GATK Unified Genotyper (v.3.5; https://gatk.broadinstitute.org), and both SNPs and indels were filtered using the GATK Variant Filtration function with proper standards (-Window 4, -filter "QD < 4.0 || FS > 60.0 || MQ < 40.0 ", -G_filter "GQ < 20"). All mutations for genes, functions, and genomic regions were annotated using ANNOVAR (Wang et al. 2010). Association analysis was performed using, ∆(SNP-Index) (Abe et al. 2012; Takagi et al. 2013), G-value (Magwene et al. 2011; Mansfeld et al. 2018), ED (Hill et al. 2013), and two-tailed Fisher’s exact test (Fisher et al. 1922) values; the overlapping interval of the four methods was considered as the final QTL interval.
Development of SNP Markers and Narrowing Candidate Interval
KASP markers were designed using Premier 5.0 (Additional file 1: Table S4), and markers that were polymorphic between parents were used to validate BSA-seq results and construct a linkage map, by genotyping the 638 progeny individuals, and to narrow the potential candidates using the Inclusive Composite Interval Mapping (ICIM) module of QTL IciMapping (v.4.2; http://www.isbreeding.net). The Logarithm of the odds (LOD) score threshold for confirming a significant QTL was established using a permutation test with 1000 replicates and a significant level of P < 0.01.
Knockout Plant Construction
CRISPR/Cas9 gene-editing vector construction was conducted as described by Li et al. (2017). Two target sequences, including Point accepted mutation (PAM) (GGCAAGGGAGGGAAGGGTCTCGG / CGTCTACGCCCTCAAGCGCCAGG) were selected within the target genes, and the targeting specificity was confirmed using a BLAST search against the rice genome (Hsu et al. 2013). Genomic DNA was extracted from these knockout lines; and after PCR amplification, the designed target site amplicon (300–500 bp) was sequenced directly and identified using the Degenerate Sequence Decoding method (Ma et al. 2015). Knockout lines were confirmed by PCR sequencing, with the primers 5’-CCCAATACAGATCAACCAAA-3’ and 5’-AGAACTGAACTACAGCAAGT-3’. The knockout lines were potted (inner diameter 30 cm), along with the control (WT) variety ‘Dongnong 430’, in the spring of 2021 at Northeast Agricultural University (Harbin, China), and upon reaching maturity, the height of each line (n = 5) was evaluated.
Statistical Analysis
Differences between parent and progeny plant height were detected using SPSS18.0 (IBM, Armonk, NY, USA). Data represent means ± standard deviations. Graphs were drawn using edgeR (http://www.r-project.org/) and Origin 2018 (OriginLab, Northampton, MA, USA).
Results
Screening and Evaluation of Plant Height
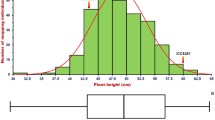
Of the two parental varieties, ‘DF114’ plants were shorter than ‘LY11’ plants (Fig. 1A, B), and the heights of the 628 F2:3 progenies (Fig. 1C) ranged from 82.5 to 117.3 cm, with the 30 shortest and tallest progenies assigned to the L-pool and H-pool, respectively, for DNA resequencing. In addition, both the skewness and kurtosis associated with plant height in the F2:3 population were close to 1 (Additional file 1: Table S1), which indicated that the data were suitable for QTL analysis.

Phenotype analysis of plant height in the progeny of a cross between ‘DF114’ and ‘LY11’. A Mature ‘DF114’ plant. B Mature ‘LY11’ plant. C Distribution of plant height among 638 mapping individuals of F2:3 population
Whole-Genome Resequencing and BSA-Seq Analysis
The mean coverage depth for the parents and the two pools was 50 × , and comparison of the sequences to the ‘Nipponbare’ reference genome resulted in the identification of 801,247 SNPs and 133,510 indels, which were reduced to 277,729 SNPs and 45,142 indels by trimming and filtering (Additional file 1: Table S2). A total of 322,871 high-quality SNPs/indels that were homozygous in each parent and polymorphic between the parents were then selected for BSA-seq analysis, using ∆(SNP-Index) values (Fig. 2A), Euclidean distance (ED) values (Fig. 2B), and G-values (Fig. 2C), as well as Fisher’s exact test P-values (Fig. 2D), were used to identify candidate plant height-related QTL regions (Table 1). Two significant (P < 0.01) peaks in the ∆(SNP-Index) distribution spanned 12.14-Mb (4.78–16.92 Mb) and 5.98-Mb (11.82–17.80 Mb) intervals (qPH7 and qPH9) on chromosomes 7 and 9, respectively, and significant peaks in G-value distribution completely covered the results of ∆(SNP-Index) analysis, whereas those in the ED and Fisher’s exact test P-value distributions completely covered the interval on Chromosome 9 alone. Furthermore, only the region identified in the Fisher’s exact test P-value distribution (2.02 Mb of qPH9) was included in the intersection of genome regions identified by all four methods. It is also worth noting that, in the Δ(SNP-Index) and ED value distributions, the peak values observed for qPH9 were higher than those observed for qPH7 (Table 1), which indicated a greater difference in the allele ratio between the two mixed pools. Therefore, qPH9 was considered to be a more significant target for mining candidate plant height genes. There is a known panicle gene DEP1 in the qPH9 interval, and the DEP1 genome sequence analysis shows that there is no difference between ‘DN114’ and ‘LY11’ (Additional file 2: Fig. S1).

Quantitative trait locus (QTL) analysis of rice plant height at maturity using 4 QTL-seq methods. A Manhattan plot showing the distribution of Δ(SNP-index) on chromosomes. B Manhattan plot showing the distribution of Euclidean distance (ED2) on chromosomes. C Manhattan plot showing the distribution of G-value on chromosomes. D Manhattan plot showing the distribution of log-transformed Fisher’s exact test P-value distribution, –log10(p) on chromosomes. Blue and red lines represent 95 and 99% confidence intervals, respectively, and black lines represent mean values of the 4 algorithms, which were drawn using sliding window analysis. Numbers on the horizontal coordinates represent chromosome numbers
Fine Mapping of qPH9
The qPH9 coding region harbored 26 SNPs and 14 indels (Additional file 1: Table S3). The analysis of 13 highly credible SNPs (Additional file 1: Table S4) in the 638 progenies, using a LOD score of 3.0 as a threshold for consecutive occurrence, yielded a qPH9 linkage map (Fig. 3A). The qPH9 locus was simultaneously linked to plant height and anchored to the 126 kb interval between 15,803,211 bp and 15,929,211 bp (Fig. 3B, Additional file 1: Table S5), which explained 20.50% of the phenotypic variation in plant height, and the peak LOD score was 32.95 (Table 2). The positive qPH9 allele was contributed by the ‘DF114’parent.

Fine mapping of the quantitative trait locus (qPH9) associated with rice plant height at maturity. A Detection of qPH9 by ICIM module of QTL IciMapping 4.2. The orange bar with vertical black lines and labels represents the linkage map and Kompetitive allele-specific PCR (KASP) markers. B Putative plant height genes identified at qPH9 using annotation information from the ‘Nipponbare’ reference genome (http://plants.ensembl.org/index.html/)
Putative Candidate Genes for qPH9
Annotation of the 126 kb interval, using annotation information of the ‘Nipponbare’ reference genome (http://plants.ensembl.org/index.html/), resulted in the identification of 15 candidate genes (Fig. 3B). These genes include 4 hypothetical proteins, 2 non-coding proteins, 2 C2H2 transcription factor family proteins, 2 Glycosyl transferase proteins, 1 Cold-regulated protein, 1 AAA-type ATPase family protein, 1 Histone H4 protein, 1 Pectin methylesterase and 1 Senescence-associated protein (Additional file 1: Table S6). The resequencing data (Additional file 1: Table S3) indicated that the difference between ‘DN114’ and ‘LY11’ were in the 15,803,211 bp and 15,929,211 position of Chromosome 9, which was within the range of 126 kb. Among them, SNP (9:15,916,244) is a non-synonymous mutation in the CDs region of Os09g0433600. In addition, the genome sequence of Os09g0433600 also showed that, there were only SNP differences at 9:15,916,244 between Os09g0433600 of ‘DN114’ and ‘LY11’ (Additional file 3: Fig. S2). Therefore, we believe that Os09g0433600 is a candidate gene for qPH9, and named OsPH9.
Significant Association SNP of OsPH9 with Plant Height by RFGB Database
With RFGB Database analysis (Fig. 4; Additional file 1: Table S7), 8 SNPs were obtained in the promoter region and CDs region of OsPH9, which were 15,915,682 (C > A/M), 15,915,683 (A > G/R), 15,915,701 (T > G/K), 15,915,717C (C > T) and 15,915,782 (T > C) in the promoter region, and 15,916,129 (C > T), 15,916,186 (C > A/M) and 15,916,244 (T > C/-) in the CDs region, respectively. A total of 11 haplotypes were obtained based on the different combinations of 8 SNPs. As shown in Fig. 4B, the plant height difference between haplotypes is significant, which indicates that the sequence difference of OsPH9 is closely related to the change in plant height. Among them, 15,916,244 (T > C) is consistent with resequencing results (Additional file 1: Table S3), indicating that 15,916,244 (T > C) may be the main reason for the difference in plant height between ‘DF114’ and ‘LY11’. We then referred to the data from the 3010 Rice Genomes Project and found that 15,916,244C mainly exists in the genotype of japonica, its allele frequency was 2.82% in japonica; however, it did not occur in indica. It could thus be inferred that 15,916,244C is a rare natural variation (Additional file 1: Table S8). Therefore, it can be used as a special functional variation in ‘DF114’ to improve the plant height of rice varieties.

Haplotype and sequence analysis. A Haplotype analysis of Os09g0433600, B Plant height differences of different haplotypes
Knockout of OsPH9 using CRISPR/Cas9 System
Functional analysis of the H4 histone coding gene Os09g0433600 (OsPH9), was conducted using the CRISPR/Cas9-generated osph9-mutant lines (A467-3 and A467-12) in which the motifs GGG and GC were deleted at the 26 and 247 bp of the CDs region, respectively (Fig. 5A). This revealed that the loss-of-function of OsZOS9-12 resulted in reduced height and node length (Fig. 5B,C), with the mature plant height of lines A467-3 and A467-12 being reduced by 7.96 and 8.99%, respectively, when compared to the wild-type plants (Fig. 5D).

Functional analysis of the H4 histone coding gene Os09g0433600 (OsPH9) in rice. A DNA sequences of Os09g0433600 in ‘Dongnong 430’ (WT) and knockout lines (A467-3 and A467-12). B Mature plant height phenotypes of WT and knockout mutant lines. C Stem node lengths of WT and knockout mutant lines. D Mature plant heights of WT and knockout mutant lines (**P < 0.01; Student’s t-test)
Discussion
Plant height has been a major breeding target in rice, due to the importance of lodging resistance and mechanized harvesting requirements. Many dwarf and semi-dwarf genes have been identified in rice. For example, mutations in OsBR6ox, which encodes a key enzyme in brassinosteroid biosynthesis, and in genes that regulate GA synthesis (e.g., SD1, D18, D35) leading to varying degrees of dwarfing (Hong et al. 2002; Monna et al. 2002; Tong et al. 2014; Wu et al. 2014). However, the continuous mining of novel height-related genes is essential for developing an ideal rice plant type and for improving rice yield. Thus, the aim of the present study was to investigate the genetic basis for short plant height in ‘DF114’ rice, thereby establishing a theoretical foundation for future plant type improvement via integrated BSA-seq and linkage mapping analysis.
Along with the recent development and application of high-throughput sequencing, high-resolution mass spectrometry, and information processing, BSA-seq has gradually matured. Its accuracy and cost have improved significantly. The combination of traditional mapping and BSA-seq can effectively and quickly reduce the major QTL interval (Takagi et al. 2015L, Lei et al. 2020; Yang et al. 2021). For instance, Zhang et al. (2021) used BSA-seq to identify a major QTL (qPH8) in a large rice population with an effect equivalent to that of SD1. In the present study, BSA-seq was used to identify a major QTL (qPH9), and regional linkage mapping analysis was used to rapidly reduce the implicated interval, which was responsible for 20.50% of plant height variation, from 2.02 Mb to 126 kb. Interestingly, previous studies have identified plant height loci that are either near or inclusive of qPH9, and qPH9 also overlaps with qSL28-9 and qPH9-1 (RZ422-RZ12), which are 3.49 and 7.62 Mb intervals linked to plant height (Huang et al. 1996; MacMillan et al. 2006). Meanwhile, ph9.1 (Marri et al. 2005) and qPH9a (Li et al. 2003), which are also QTLs associated with mature plant height, were mapped to locations 39.66 kb and 12.44 kb away from qPH9. However, of all the QTLs on rice Chromosome 9 related to plant height, qPH9 appears to be the most influential.
During the past decade, genotype datasets for many rice accessions have been released and used to identify several loci associated with important agronomic traits (Li et al. 2020; Peng et al. 2020). Several large-scale rice collections with sequence and phenotype data have provided valuable materials and knowledge for rice research and breeding projects (Li et al. 2020; Crowell et al. 2016; Zhao et al. 2018; Dong et al. 2018). Sha et al. (2021) used MBKbase-rice (www.mbkbase.org/rice) and 295 japonica cultivars from northeast China to analyze the allelic variation in SD1, which further enriched the allele types related to SD1 plant height. This study found that there are multiple haplotypes in OsPH9 in the 3 k germplasm resources and are closely related to rice plant height. It can be inferred that the mutation of OsPH9 in CDs region 15,916,244 (T > C) is the main cause of decrease in ‘DF114’ plant height.
Histones are the basic structural proteins of chromosomes. These are alkaline in nature due to the abundance of basic amino acids Arg and Lys, thus binding tightly to the acidic DNA. Histones consist of five components with molecular masses ranging from 11 to 23 ku, which are called H1, H3, H2A, H2B, and H4 according to their molecular weights from large to small. The gene, OsPH9, is a H4 histone coding gene. Histone encoding genes are conservative, and among the distant species, the amino acid sequences of the four histones (H2A, H2B, H3, H4) are very similar, especially H3 and H4 histones (Mariño-Ramírez et al. 2005). Previous studies have shown that different variants of histones are closely related to the growth and development of organisms (Zhou et al. 2015; Nacev et al. 2019). The mutation of yH3 H113 in yeast caused a significant decrease in the affinity of yH3-H4 tetramer for copper ions (Attar et al. 2020). Du et al. (2020) showed OsChz1 in rice, as a common molecular chaperone of H2A-H2B and H2A.Z-H2B, which dynamically regulates the distribution of higher eukaryotes by regulating the density of nucleosomes and the distribution of histone variants H2A.Z. Chromatin structure, which in turn regulates its developmental process. In order to further prove the function of OsPH9 in plant height, the CRISPR/Cas9 was carried out, and the results showed that the plant height of osph9-mutant lines were significantly lower than the WT. These results have important significance in the field of rice plant type/plant height improvement breeding.
Conclusion
Investigating the genetic basis of plant height is important for breeding purposes. In the present study, BSA-seq and linkage-map analysis were used to identify a QTL (i.e., qPH9) related with the height of a mature rice plant. Experimental verification by haplotype and sequence analysis confirmed OsPH9 as a candidate gene of the qPH9 region, and the function of OsPH9 was further verified using gene-editing technology. Although further work is needed to elucidate the mechanism underlying the role of OsPH9 in plant height, the present study provides resources for breeding programs aimed at improving rice plant height.
Data Availability Statement
The BSA-seq data for this study can be found in the National Center for Biotechnology Information Sequence Read Archive under the accession numbers SSRR13306959, SRR13306960, SRR13306961, and SRR13306962 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA687818).
Abbreviations
- ABA:
-
Abscisic acid
- Add:
-
Additive effects
- BSA:
-
Bulked segregant analysis
- CDS:
-
Coding sequence
- Chr:
-
Chromosome
- DF114:
-
Dongfu114
- DN430:
-
Dongnong 430
- ED:
-
Euclidian distance
- GA:
-
Gibberellic acid
- KASP:
-
Kompetitive allele-specific PCR
- LY11:
-
Longyang11
- LOD:
-
Logarithm of the odds
- PVE:
-
Phenotypic variation explained
- QTL:
-
Quantitative trait locus
- RFGB:
-
Rice functional genomics and breeding
- SNP:
-
Single nucleotide polymorphisms
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30(2):174–178. https://doi:https://doi.org/10.1038/nbt.2095
Attar N, Campos OA, Vogelauer M, Cheng C, Xue Y, Schmollinger S, Salwinski L, Mallipeddi NV, Boone BA, Yen L, Yang S, Zikovich S, Dardine J, Carey MF, Merchant SS, Kurdistani SK (2020) The histone H3–H4 tetramer is a copper reductase enzyme. Science 369(6499):59–64. https://doi.org/10.1126/science.aba8740
Chen WF, Xu ZJ, Zhang WZ, Zhang LB, Yang SR (2001) Creation of new plant type and breeding rice for super high yield. Acta Agron Sin 27(5):665–673
Crowell S, Korniliev P, Falcão A, Ismail A, Gregorio G, Mezey J, McCouch S (2016) Genome-wide association and high-resolution phenotyping link Oryza sativa panicle traits to numerous trait-specific QTL clusters. Nat Commun 7:10527. https://doi.org/10.1038/ncomms10527
Dong H, Zhao H, Li S, Han Z, Hu G, Liu C, Yang G, Wang G, Xie W, Xing Y (2018) Genome-wide association studies reveal that members of bHLH subfamily 16 share a conserved function in regulating flag leaf angle in rice (Oryza sativa). PLoS Genet 14(4):e1007323. https://doi.org/10.1371/journal.pgen.1007323
Du K, Luo Q, Yin L, Wu J, Liu Y, Gan J, Dong A, Shen WH (2020) OsChz1 acts as a histone chaperone in modulating chromatin organization and genome function in rice. Nat Commun 11(1):5717. https://doi.org/10.1038/s41467-020-19586-z
Fisher RA (1922) On the interpretation of χ 2 from contingency tables and the calculation of P. J Roy Stat Soc 85(1):87–94
Giovannoni JJ, Wing RA, Ganal MW, Tanksley SD (1991) Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res 19(23):6553–6558. https://doi.org/10.1093/nar/19.23.6553
Hedden P (2003) The genes of the Green Revolution. Trends Genet 19:5–9. https://doi.org/10.1016/s0168-9525(02)00009-4
Hill JT, Demarest BL, Bisgrove BW, Gorsi B, Su YC, Yost HJ (2013) MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res 23(4):687–697. https://doi.org/10.1101/gr.146936.112
Hong Z, Ueguchi-Tanaka M, Shimizu-Sato S, Inukai Y, Fujioka S, Shimada Y, Takatsuto S, Agetsuma M, Yoshida S, Watanabe Y, Uozu S, Kitano H, Ashikari M, Matsuoka M (2002) Loss-of-function of a rice brassinosteroid biosynthetic enzyme C-6 oxidase prevents the organized arrangement and polar elongation of cells in the leaves and stem. Plant J 32(4):495–508. https://doi.org/10.1046/j.1365-313x.2002.01438.x
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9):827–832. https://doi.org/10.1038/nbt.2647
Huang P, Jiang H, Zhu C, Barry K, Jenkins J, Sandor L, Schmutz J, Box MS, Kellogg EA, Brutnell TP (2017) Sparse panicle1 is required for inflorescence development in Setaria viridis and maize. Nature Plants 3:17054. https://doi.org/10.1038/nplants.2017.54
Huang N, Courtois B, Khush GS, Lin HX, Wang GL, Wu P, Zheng KG (1996) Association of quantitative trait loci for plant height with major dwarfing genes in rice. Heredity 77:130–137
Hubbard KE, Nishimura N, Hitomi K, Getzoff ED, Schroeder JI (2010) Early abscisic acid signal transduction mechanisms: newly discovered components and newly emerging questions. Genes Dev 24(16):1695–1708. https://doi.org/10.1101/gad.1953910
Kadambari G, Vemireddy LR, Srividhya A, Nagireddy R, Jena SS, Gandikota M, Patil S, Veeraghattapu R, Deborah D, Reddy GE, Shake M, Dasari A, Ramanarao PV, Durgarani CV, Neeraja CN, Siddiq EA, Sheshumadhav M (2018) QTL-Seq-based genetic analysis identifies a major genomic region governing dwarfness in rice (Oryza sativa L.). Plant Cell Rep 37(4) 677–687. https://doi.org/10.1007/s00299-018-2260-2
Kumar S, Agarwal S, Ranvijay (2019). Fast and memory efficient approach for mapping NGS reads to a reference genome. J Bioinform Comput Biol 17(2) 1950008
Lei L, Zheng H, Bi Y, Yang L, Liu H, Wang J, Sun J, Zhao H, Li X, Li J, Lai Y, Zou D (2020) Identification of a major QTL and candidate gene analysis of salt tolerance at the bud burst stage in rice (Oryza sativa L.) using QTL-Seq and RNA-Seq. Rice 10 13(1):55. https://doi:https://doi.org/10.1186/s12284-020-00416-1
Li H, Hu B, Chu C (2017) Nitrogen use efficiency in crops: lessons from Arabidopsis and rice. J Exp Bot 68(10):2477–2488. https://doi.org/10.1093/jxb/erx101
Li X, Chen Z, Zhang G, Lu H, Qin P, Qi M, Yu Y, Jiao B, Zhao X, Gao Q, Wang H, Wu Y, Ma J, Zhang L, Wang Y, Deng L, Yao S, Cheng Z, Yu D, Zhu L, Xue Y, Chu C, Li A, Li S, Liang C (2020) Analysis of genetic architecture and favorable allele usage of agronomic traits in a large collection of Chinese rice accessions. Science China Life Sciences 63(11):1688–1702. https://doi.org/10.1007/s11427-019-1682-6
Li ZK, Yu SB, Lafitte HR, Huang N, Courtois B, Hittalmani S, Vijayakumar CH, Liu GF, Wang GC, Shashidhar HE, Zhuang JY, Zheng KL, Singh VP, Sidhu JS, Srivantaneeyakul S, Khush GS (2003) QTL x environment interactions in rice. I. heading date and plant height. Theor Appl Genet 108(1) 141–153. https://doi.org/10.1007/s00122-003-1401-2
Lin Q, Zhang Z, Wu F, Feng M, Sun Y, Chen W, Cheng Z, Zhang X, Ren Y, Lei C, Zhu S, Wang J, Zhao Z, Guo X, Wang H, Wan J (2020) The APC/CTE E3 ubiquitin ligase complex mediates the antagonistic regulation of root growth and tillering by ABA and GA. Plant Cell 32(6):1973–1987. https://doi:https://doi.org/10.1105/tpc.20.00101
Liu MM, Shi ZY, Zhang XH, Wang MX, Zhang L, Zheng KZ, Liu JY, Hu XM, Di CR, Qian Q, He ZH, Yang DL (2019) Inducible overexpression of ideal plant architecture1 improves both yield and disease resistance in rice. Nat Plants 5:389–400. https://doi.org/10.1038/s41477-019-0383-2
Liu S, Hua L, Dong S, Chen H, Zhu X, Jiang J, Zhang F, Li Y, Fang X, Chen F (2015) OsMAPK6 a mitogen-activated protein kinase influences rice grain size and biomass production. Plant J 84(4):672–681. https://doi.org/10.1111/tpj.13025
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, Wang B, Yang Z, Li H, Lin Y, Xie Y, Shen R, Chen S, Wang Z, Chen Y, Guo J, Chen L, Zhao X, Dong Z, Liu YG (2015) A robust CRISPR/Cas9 system for convenient high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8(8):1274–1284. https://doi.org/10.1016/j.molp.2015.04.007
MacMillan K, Emrich K, Piepho HP, Mullins CE, Price AH (2006) Assessing the importance of genotype x environment interaction for root traits in rice using a mapping population II: conventional QTL analysis. Theor Appl Genet 113(5):953–964. https://doi.org/10.1007/s00122-006-0357-4
Magwene PM, Willis JH, Kelly JK (2011) The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput Biol 7(11):e1002255. https://doi.org/10.1371/journal.pcbi.1002255
Mansfeld BN, Grumet R (2018) QTLseqr: an R Package for Bulk Segregant analysis with next-generation sequencing. Plant Genome 11(2):10.3835. https://doi.org/10.3835/plantgenome2018.01.0006
Mariño-Ramírez L, Kann MG, Shoemaker BA, Landsman D (2005) Histone structure and nucleosome stability. Expert Rev Proteomics 2(5):719–729. https://doi.org/10.1586/14789450.2.5.719
Marri PR, Sarla N, Reddy LV, Siddiq EA (2005) Identification and mapping of yield and yield related QTLs from an Indian accession of Oryza rufipogon. BMC Genet 6:33. https://doi.org/10.1186/1471-2156-6-33
Matsumoto T, Wu JZ, Kanamori H, Katayose Y, Fujisawa M, Namiki N, Mizuno H, Yamamoto K, Antonio BA, Baba T (2005) The map-based sequence of the rice genome. Nature 436(7052):793–800. https://doi.org/10.1038/nature03895
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA 88(21):9828–9832. https://doi.org/10.1073/pnas.88.21.9828
Monna L, Kitazawa N, Yoshino R, Suzuki J, Masuda H, Maehara Y, Tanji M, Sato M, Nasu S, Minobe Y (2002) Positional cloning of rice semidwarfing gene sd-1: rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res 9(1):11–17. https://doi.org/10.1093/dnares/9.1.11
Monna L, Miyao A, Zhong HS, Sasaki T, Minobe Y (1995) Screening of RAPD markers linked to the photoperiod-sensitivity gene in rice chromosome 6 using bulked segregant analysis. DNA Res 2(3):101–106. https://doi.org/10.1093/dnares/2.3.101
Nacev BA, Feng L, Bagert JD, Lemiesz AE, Gao J, Soshnev AA, Kundra R, Schultz N, Muir TW, Allis CD (2019) The expanding landscape of “oncohistone” mutations in human cancers. Nature 567(7749):473–478. https://doi.org/10.1038/s41586-019-1038-1
Peng J, Richards DE, Hartley NM, Murphy GP, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AJ, Pelica F, Sudhakar D, Christou P, Snape JW, Gale MD, Harberd NP (1999) Green revolution’ genes encode mutant gibberellin response modulators. Nature 400(6741):256–261. https://doi.org/10.1038/22307
Peng H, Wang K, Chen Z, Cao Y, Gao Q, Li Y, Li X, Lu H, Du H, Lu M, Yang X, Liang C (2020) MBKbase for rice: an integrated omics knowledgebase for molecular breeding in rice. Nucleic Acids Res 48(D1):D1085–D1092. https://doi.org/10.1093/nar/gkz921
Ramirez-Gonzalez RH, Segovia V, Bird N, Fenwick P, Holdgate S, Berry S, Jack P, Caccamo M, Uauy C (2015) RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol J 13(5):613–624. https://doi.org/10.1111/pbi.12281
Sakamoto T, Miura K, Itoh H, Tatsumi T, Ueguchi-Tanaka M, Ishiyama K, Kobayashi M, Agrawal GK, Takeda S, Abe K, Miyao A, Hirochika H, Kitano H, Ashikari M, Matsuoka M (2004) An overview of gibberellin metabolism enzyme genes and their related mutants in rice. Plant Physiol 134(4):1642–1653. https://doi.org/10.1104/pp.103.033696
Sha H, Liu H, Zhao G, Han Z, Chang H, Wang J, Zheng H, Zhang J, Yu Y, Liu Y, Zou D, Nie S, Fang J (2021) Elite sd1 alleles in japonica rice and their breeding applications in northeast China. Crop J. https://doi.org/10.1016/j.cj.2021.05.005
Shen AH, Luo HB, Deng ZP, Zhou ZJ (2014) Recent advances in brassinosteroid signaling in rice. Zhejiang Nongye Xuebao (acta Agriculturae Zhejiangensis) 26(5):1399–1404
Song S, Dai X, Zhang WH (2012) A rice F-box gene OsFbx352 is involved in glucose-delayed seed germination in rice. J Exp Bot 63(15):5559–5568. https://doi.org/10.1093/jxb/ers206
Spielmeyer W, Ellis MH, Chandler PM (2002) Semidwarf (sd-1) “green revolution” rice contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci USA 99(13):9043–9048. https://doi.org/10.1073/pnas.132266399
Sun TP (2010) Gibberellin-GID1-DELLA: a pivotal regulatory module for plant growth and development. Plant Physiol 154(2):567–570. https://doi.org/10.1104/pp.110.161554
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74(1):174–183. https://doi.org/10.1111/tpj.12105
Tan L, Li X, Liu F, Sun X, Li C, Zhu Z, Fu Y, Cai H, Wang X, Xie D, Sun C (2008) Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 40(11):1360–1364. https://doi.org/10.1038/ng.197
Tong H, Xiao Y, Liu D, Gao S, Liu L, Yin Y, Jin Y, Qian Q, Chu C (2014) Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell 26(11):4376–4393. https://doi.org/10.1105/tpc.114.132092
Wambugu P, Ndjiondjop MN, Furtado A, Henry R (2018) Sequencing of bulks of segregants allows dissection of genetic control of amylose content in rice. Plant Biotechnol J 16(1):100–110. https://doi.org/10.1111/pbi.12752
Wang F, Han T, Song Q, Ye W, Song X, Chu J, Li J, Chen ZJ (2020) The rice circadian clock regulates tiller growth and panicle development through strigolactone signaling and sugar sensing. Plant Cell 32(10):3124–3138. https://doi.org/10.1105/tpc.20.00289
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. https://doi.org/10.1093/nar/gkq603
Wei X, Zhou H, Xie D, Li J, Yang M, Chang T, Wang D, Hu L, Xie G, Wang J, Wang L (2021) Genome-wide association study in rice revealed a novel gene in determining plant height and stem development by encoding a WRKY transcription factor. Int J Mol Sci 22(15):8192. https://doi.org/10.3390/ijms22158192
Wen J, Jiang F, Weng Y, Sun M, Shi X, Zhou Y, Yu L, Wu Z (2019) Identification of heat-tolerance QTLs and high-temperature stress-responsive genes through conventional QTL mapping QTL-seq and RNA-seq in tomato. BMC Plant Biol 19(1):398. https://doi.org/10.1186/s12870-019-2008-3
Wu J, Zhu C, Pang J, Zhang X, Yang C, Xia G, Tian Y, He C (2014) OsLOL1 a C2C2-type zinc finger protein interacts with OsbZIP58 to promote seed germination through the modulation of gibberellin biosynthesis in Oryza sativa. Plant J 80(6):1118–1130. https://doi.org/10.1111/tpj.12714
Wu K, Wang S, Song W, Zhang J, Wang Y, Liu Q, Yu J, Ye Y, Li S, Chen J, Zhao Y, Wang J, Wu X, Wang M, Zhang Y, Liu B, Wu Y, Harberd NP, Fu X (2020) Enhanced sustainable green revolution yield via nitrogen-responsive chromatin modulation in rice. Science (New York N.Y.) 367(6478):eaaz2046. https://doi.org/10.1126/science.aaz2046
Yaish MW, El-Kereamy A, Zhu T, Beatty PH, Good AG, Bi YM, Rothstein SJ (2010) The APETALA-2-like transcription factor OsAP2-39 controls key interactions between abscisic acid and gibberellin in rice. PLoS Genet 6(9):e1001098. https://doi.org/10.1371/journal.pgen.1001098
Yang L, Wang J, Han Z, Lei L, Liu HL, Zheng H, Xin W, Zou D (2021) Combining QTL-seq and linkage mapping to fine map a candidate gene in qCTS6 for cold tolerance at the seedling stage in rice. BMC Plant Biol 21(1):278. https://doi.org/10.1186/s12870-021-03076-5
Zegeye WA, Zhang Y, Cao L, Cheng S (2018) Whole genome resequencing from bulked populations as a rapid QTL and gene identification method in rice. Int J Mol Sci 19(12):4000. https://doi.org/10.3390/ijms19124000
Zhang B, Qi F, Hu G, Yang Y, Zhang L, Meng J, Han Z, Zhou X, Haiyang Liu H, Ayaad M, Xing, Y (2021) Bsa-seq-based identification of a major additive plant height qtl with an effect equivalent to that of semi-dwarf 1 in a large rice f2 population. Crop J (42). https://doi.org/10.1016/j.cj.2020.11.011
Zhang G, Angeles ER, Abenes ML, Khush GS, Huang N (1996) RAPD and RFLP mapping of the bacterial blight resistance gene xa-13 in rice. Theor Appl Genet 93(1–2):65–70. https://doi.org/10.1007/BF00225728
Zhang Q, Shen BZ, Dai XK, Mei MH, Saghai A, Li ZB (1994) Using bulked extremes and recessive class to map genes for photoperiod-sensitive genic male sterility in rice. Proc Natl Acad Sci USA 91(18):8675–8679. https://doi.org/10.1073/pnas.91.18.8675
Zhang L, Yu H, Ma B, Liu G, Wang J, Wang J, Gao R, Li J, Liu J, Xu J, Zhang Y, Li Q, Huang X, Xu J, Li J, Qian Q, Han B, He Z, Li J (2017) A natural tandem array alleviates epigenetic repression of IPA1 and leads to superior yielding rice. Nature Communications 8:14789. https://doi.org/10.1038/ncomms14789
Zhao Q, Feng Q, Lu H, Li Y, Wang A, Tian Q, Zhan Q, Lu Y, Zhang L, Huang T, Wang Y, Fan D, Zhao Y, Wang Z, Zhou C, Chen J, Zhu C, Li W, Weng Q, Xu Q, Wang ZX, Wei X, Han B, Huang X (2018) Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat Genet 50(2):278–284. https://doi.org/10.1038/s41588-018-0041-z
Zhou W, Zhu Y, Dong A, Shen WH (2015) Histone H2A/H2B chaperones: from molecules to chromatin-based functions in plant growth and development. Plant J 83(1):78–95. https://doi.org/10.1111/tpj.12830
Zou C, Wang P, Xu Y (2016) Bulked sample analysis in genetics genomics and crop improvement. Plant Biotechnol J 14(10):1941–1955. https://doi.org/10.1111/pbi.12559
Acknowledgements
Thank for the funding form the major science and technology project of Heilongjiang Province, China (grant no. 2020ZX16B010).
Funding
This research was financially supported by the major science and technology project of Heilongjiang Province, China (Grant No. 2020ZX16B010), National Natural Science Foundation of China (Grant No. 32101748).
Author information
Authors and Affiliations
Contributions
W.X. and D.Z. conceived and designed the research; W.X., L.Y., and T.M. participated in data analysis; J.W., H.Z., and W.L. performed material development, sample preparation, and data analysis; W.X. wrote the manuscript; H.Z and D.Z. corrected the manuscript; and the final manuscript was read and approved by all of the authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflicts of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table S1: Phenotypic variation of parents and F2:3 lines. Table S2: Number of single nucleotide polymorphisms (SNPs) and Indels detected in the samples. Table S3: Statistics of variation in the 2.02 Mb interval of qPH9 based on resequencing data. Table S4: Primers used for the 13 KASP markers. Table S5: Further mapping information for qPH9 using the 13 KSAP markers. Table S6: Annotated genes within the 126-Kb interval. Table S7: Haplotype and plant height difference of Os09g0433600. Table S8: Haplotype and plant height difference of chr9:15916244 T. Table S9: Information on 26 germplasm resources of Hap6 haplotypes ofOs09g0433600.
Additional file 2
. Figure S1: The sequence information of DEP1 in ‘Dongfu 114’ and ‘Longyang 11’.
Additional file 3
. Figure S2: The sequence information of Os09g0433600 in ‘Dongfu 114’ and ‘Longyang 11’.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xin, W., Liu, H., Yang, L. et al. BSA-Seq and Fine Linkage Mapping for the Identification of a Novel Locus (qPH9) for Mature Plant Height in Rice (Oryza sativa). Rice 15, 26 (2022). https://doi.org/10.1186/s12284-022-00576-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-022-00576-2