
Abstract
Titanium-doped silicon clusters anions, TimSi −n (m = 1‒2, n = 14‒20), have been investigated by photoelectron spectroscopy and density functional theory (DFT) calculations. Low-energy structures of TimSi −n clusters have been globally searched using a genetic algorithm combined with DFT calculations. The electronic density of states and vertical detachment energies have been computed at the HSE06/aug-cc-pVDZ level and compared to the experimental data. Excellent agreement is found between theory and experiment especially in case of the singly doped clusters. In general, clusters with size m + n ≤ 17 prefer cage structures, while larger sized clusters evolve on a quasi-fullerene Ti@Si14 structural motif. Natural population analysis reveals that the Ti atoms possess negative charges and thus act as electron acceptors. The calculated binding energies and HOMO–LUMO gaps show that the clusters with cage structures have significantly higher stability, particularly Ti1Si16− and Ti2Si15−. One reason is that neutral Ti2Si15 exhibits a closed-shell electronic structure as a superatom, like Ti1Si16.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The continuous miniaturization of silicon-based electronic devices has stimulated intensive explorations of the electronic properties of silicon-based nanostructures. Silicon nanoclusters have attracted enormous attention in the past three decades as promising building blocks for self-assembled structures in the microelectronics industry. Their physical and chemical properties can be tailored by cluster size and geometry [1,2,3,4,5,6]. However, with the preferential sp3 bonding nature, Si clusters inevitably have surface dangling bonds, which makes it difficult to form very stable structures [7]. Doping metal atoms into Si clusters is an effective strategy to stabilize the cluster structure and also tailor the cluster properties [1]. For instance, the cage-like cluster Si16 encapsulating a Ti atom was demonstrated to be one of the most stable Si-based clusters [8,9,10]. It was first predicted theoretically in 2001 by Kumar and Kawazoe [4], and recently synthesized by Nakajima’s group on a macroscopic scale based on a clean dry-physical process [11]. Ti atom doping induces a large HOMO–LUMO gap of 1.90 eV [12], giving the clusters potential for applications in electronics, optoelectronics and optics. Si clusters doped by other metal atoms such as Sc, V, Cr, Zr, and Hf have also been theoretically studied and produced in the laboratory [12,13,14,15].
To date, Si clusters doped by a single Ti atom (Ti1@Sin) have been extensively investigated, including their atomic structures [16,17,18,19,20], stability [21, 22], electronic, catalytic [23] and optical properties for a wide range of cluster size [6, 9, 24,25,26]. The mass spectra showed that neutral Ti1Si16 is a “magic number” with high abundance [12], while in the mass spectra of anions both Ti1Si15 and Ti1Si16 clusters exhibit high intensities [27]. Koyasu et al. measured the HOMO–LUMO gap of the Ti1Si16 cluster to be ~ 1.90 eV by photoelectron spectroscopy [12, 28]. In addition, mass spectrometry, photoelectron spectroscopy and adsorption reactivity toward H2O demonstrated that the neutral M1@Sin (M = Ti, Zr and Hf) clusters possess highest stability at n = 16 due to closed electronic shells and highly symmetric geometries [15, 29].
On the theoretical side, putative ground state geometries of neutral Ti1@Sin clusters have been determined. The clusters tend to form basket structures with n up to 12, while the cage structure becomes favorable for n ≥ 13. The binding energy increases with the cluster size, and attains a maximum for n = 16 [16, 17]. For Ti1@Sin clusters with n > 16, the excess Si atoms aggregate into an apical bud [18, 30]. Koyasu et al. [12] conjectured that the high stability of Ti1@Si16 is attributed to a 20-electron shell closing by parts of the valence electrons, which were assumed to behave like a nearly free electron gas inside the Si cage [31, 32]. Later, Torres et al. emphasized that all 68 valence electrons produce a shell closing [33, 34]. The bulk materials assembled from Ti1@Si16 clusters can exhibit high-temperature superconducting properties [9]. Also, a linear heterotrimer of the fullerene-like M1@Si16 (M = Sc, Ti and V) clusters was shown to have a large dipole moment [25], potentially suitable for optoelectronics such as silicon-based lasers, tagging and photoexcitation.
Despite the aforementioned achievements, the knowledge on the structural and physical properties of Ti-doped Si clusters with sizes other than n = 16 is still limited. Especially, Si clusters doped by multiple Ti atoms remain unexplored. Here we carried out density functional theory (DFT) calculations combined with experimental photoelectron spectroscopy to elucidate the structure, stability and electronic properties of mono- and bi-doped Si cluster anions TimSi −n (m = 1, 2) with sizes n = 14‒20. The low-energy isomer structures of TimSi −n were searched and the ground state structures were determined by comparing the computed photoelectron spectra with the experimental ones. The electronic structures of Ti-doped Si clusters were examined to identify the isomers with superatomic character. These theoretical and experimental results establish a comprehensive picture of the structure, stability and electronic properties of the low-energy isomers of Ti-doped Si clusters as a function of cluster size and help to identify building blocks for novel self-assembled silicon-based nanomaterials.
2 Experimental and theoretical methods
2.1 Experimental
Silicon and silicon-metal alloy clusters were produced in a magnetron sputtering gas aggregation cluster source. Silicon and metal atoms were sputtered from a 2-inch target (a sandwich of a silicon target with holes drilled into it in the sputtering region and a titanium target) into a mixture of helium and argon (flux ratio roughly 3:1) with a total pressure of around 0.5 mbar inside a liquid nitrogen cooled aggregation tube. Under these conditions Si atoms condenses into Si and Si alloy clusters with various sizes. The high density of charge carriers yielded by the magnetron discharge led to effective charging of the clusters forming both anions and cations. The clusters then expanded with the buffer gas through an adjustable iris into vacuum. A radio-frequency (RF) octupole ion guide steered the cluster anions into the next chamber where they were fed into a RF 12-pole cryogenic ion trap. In this trap, which was cooled to 80 K, the clusters were thermalized by collisions with precooled helium buffer gas with a pressure of about 10−3 mbar. Bunches of cluster anions were then extracted from the trap and introduced into a high resolution, double reflection time-of-flight mass spectrometer, where a multiwire mass gate located at the focal point of the first reflector would select a specific mass with a resolution of about m/∆m = 2000. The size-selected clusters then were reflected and rebunched again by the second reflector, and decelerated by a pulsed electric field. After that, the clusters entered into the interaction region of a magnetic bottle time-of-flight photoelectron spectrometer, where they were irradiated by a laser pulse from a KrF excimer laser. Typically, photoelectron spectra (PES) were averaged over 30,000 laser shots at a repetition rate of 100 Hz. The spectrometer was calibrated by measuring the known PES of platinum anions, thereby leading to an error of the measured binding energies of less than 30 meV. The mass spectrum ensured there is no silicon oxides contributed to the PES signal (Fig. S1 of Supporting Information).
2.2 Theoretical
The low-energy isomer structures of TimSi −n (m = 1‒2, n = 14‒20) clusters were searched independently by an unbiased comprehensive genetic algorithm (CGA) code [35], which is implemented within the DMol3 program [36]. The validity and efficiency of this CGA-DFT scheme have been well demonstrated in a series of our recent studies on Si [37, 38], B [39, 40], V–Si [13, 41], B–Si [42], Ti/Zr/Hf–Si [43], Fe–Ge [44] Cr–Ge [45] and Mo–B [46] clusters. Further details of CGA can be found in our recent review article [35]. For each cluster size, a few independent CGA searches were performed with different presumed symmetries, including the C1, C2, C3, and Cs point groups. With each specific symmetry constraint, every CGA search ran at least 3000 iterations and retained 16 members in the population. The mutation ratio was set as 30% to ensure the diversity of population. The resulting child clusters were optimized by DMol3 using the double numerical basis including d-polarization function (DND) and the Perdew–Burke–Enzerhof (PBE) functional within the generalized gradient approximation (GGA) [47, 48].
The energetic and electronic properties of the TimSi −n clusters obtained from CGA-DFT search were further calculated by the Gaussian09 program [49] using the aug-cc-pVDZ basis set [50] and HSE06 functional [51]. Our recent benchmark study [38] demonstrated that this methodology can provide accurate description of the structural and electronic properties for Sin clusters with n = 14‒20. For comparison, we also optimized the structures of all the considered isomers using the PBE0 functional [52]. For each cluster size, the relative energies of isomers predicted by these two functionals are nearly identical with a discrepancy less 0.02 eV (see Figs. S2, S3 of Supporting Information). Vibrational analysis were carried out to ensure that there is no imaginary frequency corresponding to the saddle point on potential energy surface and the zero-point-energy (ZPE) was included in the final energy. Spin multiplicities testing show that doublet for TimSi −n anions is most stable state, and singlet and triplet for neutral species may appear as most stable state.
The stability of a TimSi −n cluster can be characterized by the binding energy (Eb) defined as:
where E(TimSi −n ) is the energy of the anionic TimSi −n cluster; E(Si), E(Ti−) and E(Ti) are the energies of an individual Si atom, Ti− anion and Ti atom, respectively. The natural population analysis (NPA) [53] of TimSi −n was conducted using the natural bond orbital (NBO) 3.1 program [54, 55] implemented in the Gaussian09 package.
To quantitatively compare with the experimental results, the vertical detachment energy (VDE) was calculated as the energy required to remove an electron from the highest occupied molecular orbital (HOMO) without relaxing the atomic configuration, which corresponds to the first peak maxima of the photoelectron spectrum. Upon photoemission, the structure of the neutral TimSin cluster remains identical to that of the anion under the assumption of a vertical transition. The PES of anionic clusters were simulated using the ‘generalized Koopman’s theorem’ based on the energy levels from DFT calculations [56]. For the VDEs of deeper Kohn–Sham states, we shifted the calculated DOS globally in order to align the binding energy of the HOMO with the theoretical VDE value [57, 58]. Each line of the Kohn–Sham eigenvalue spectrum was broadened by a Gaussian function of width 0.06 eV.
3 Results and discussion
3.1 Structures and stability of TimSi −n
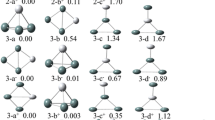
The low-energy isomers of TimSi −n anions globally searched by the CGA-DFT procedure are displayed in Figs. S2, S3 of Supporting Information. For each TimSi −n cluster system, we considered only the isomers with energies higher than the ground state by less than 0.20 eV, which are shown in Fig. 1 and denoted as n–I, n–II, n–III, and so on (from the lowest to highest energies). The relative stabilities of various isomers are given by Table 1.

Low-energy structures of TimSi −n (m = 1‒2, n = 14‒20) clusters discovered using HSE06/aug-cc-pVDZ. For each cluster size, several low-energy isomers marked as n‒I, n‒II, and n‒III. The Si and Ti atoms are shown in gold and silver colors, respectively. The cluster symmetry is given in bracket and the energy difference (in eV) relative to the ground state is provided
3.1.1 Ti1Si −n
For the mono-doped anionic Si clusters, Ti atoms are encapsulated in the center of Si cages (Fig. 1a). The smaller-sized Ti1Si −n clusters (n = 14‒16) prefer simple cage structures. In particular, the lowest-energy isomers of Ti1Si14− and Ti1Si15− adopt a bicapped pentagonal prism as the structural motif, with two and three Si atoms inserted on the waist of prism showing C2v and Cs symmetry, respectively. These are consistent with the structures reported by Reveles and Khanna [31] Both Ti1Si14− and Ti1Si15− have another isomer with energy higher than the ground state of only 0.01 and 0.09 eV, respectively. The 14–II isomer has a Si dimer hanging on a tetragon face of a hexagonal prism and therefore only has Cs symmetry, while 15–II is a basket-like structure with C2v symmetry. 14–III, 14–IV and 14–V are higher in energy than the ground state by 0.01, 0.09 and 0.12 eV, respectively, with two silicon atoms hanging on a C3v Ti@Si12 motif. The Ti1Si16− cluster has three low-lying isomers: the Frank–Kasper (FK) polyhedron structure with Td symmetry [59], a distorted FK structure with C3v symmetry [43], and a Ti@Si14 capped with a Si2 dimer with Cs symmetry [4]. The former two isomers are nearly degenerate with an energy difference of only 0.06 eV, while the latter one is higher in energy than the ground state by 0.10 eV.
A fullerene-like D3h structure of Ti@Si14 (hereafter denoted as Ti@Si14), which can be viewed as a tertruncated square bipyramid, serves as the structure motif of the larger Ti1Si −n cluster. Similar to the carbon fullerenes with pentagonal and hexagonal faces, its outer Si14 cage contains three tetragonal faces and six pentagonal faces with each Si atom being tri-coordinated. According to the Euler theorem, 18, 16, 14, 12 and 10-vertex polyhedrons could have 1, 2, 3, 4 and 5 tetragonal faces and 10, 8, 6, 4 and 2 pentagonal faces, respectively. Analogous to the isolated pentagon rule for carbon fullerene, the D3h Si14 cage can be regarded as the smallest “quasi-fullerene” obeying the “isolated tetragon rule”. For larger cluster anions (n = 17‒20), the D3h Ti@Si14 served as the structural motif is capped with various apical buds by the excess Si atoms. The Ti1Si17− cluster has two low-lying isomers with an energy difference of 0.07 eV, featured as three Si atoms capping one pentagon face or two adjacent pentagon faces of the Ti@Si14 motif, respectively. The clusters with n = 18‒19 have only one low-lying isomer within the considered energy range: Ti1Si18− with Cs symmetry can be derived by one Si atom capping a hexagonal face of the 17–II isomer; Ti1Si19− with low symmetry has a Si dimer capping a pentagon face of the Ti@Si14 motif, and three Si atoms capping the pentagon of the other side. The Ti1Si20− cluster has two low-lying isomers with an energy difference of 0.16 eV. The 20–I isomer can be derived by adding one Si atom onto the 19–I isomer, and 20–II isomer with Cs symmetry has six Si atoms capping a pentagon of the Ti@Si14 motif.
3.1.2 Ti2Si −n
For the bi-doped anionic Si clusters, one Ti atom is encapsulated in the center of Si cages, while the other Ti atom stays on the cluster surface bonding with several Si atoms (Fig. 1b). The Ti2Si14− cluster has a cage structure, while for n = 15‒20, the Ti@Si14-based structural motif starts to dominate the low-lying isomers. Specifically, the Ti2Si14− cluster has three low-lying isomers. The two lowest isomers are nearly degenerate with an energy difference of 0.08 eV. They can be viewed as a Ti@Si12 motif with the hexagonal antiprism capped by one Ti atom and one Si dimer, respectively, giving either Cs or C2v symmetry. The 14–III isomer with Cs symmetry is higher in energy by 0.11 eV than the lowest-energy one. For n = 15, the lowest-energy isomer can be derived by capping a Si–Ti dimer onto a pentagon face of the Ti@Si14− motif. The other two isomers are the FK and distorted FK polyhedrons with C3v and Cs symmetry, respectively, and are higher in energy than the ground state by 0.01 and 0.19 eV, respectively. As we will discuss in detail later, the FK isomer with C3v symmetry is a probable candidate for the structure present in the experiment. As the cluster size increases, the excess Si atoms start to evolve into a bud or sub-cage enclosing the Ti atom on the cluster surface.
For larger cluster anions (n = 17‒20), most isomers have a Ti@Si14-based structure. In particular, the Ti2Si −n clusters with n = 16, 17 and 18 can be viewed as the Ti@Si14 motif with two, three and four Si atoms capping a pentagon face surrounding the surface Ti atom, respectively. Ti2Si16−, Ti2Si17− and Ti2Si18− have two other isomers higher in energy by 0.12, 0.20 and 0.17 eV than the ground state ones, respectively. 16–II and 18–II have a TiSi2 triangle and a TiSi4 trigonal bipyramid capping a pentagon face of the Ti@Si14 motif, while 17–II is a fourteen-vertex cage capped by a TiSi3 rhombus with Cs symmetry. The Ti2Si19− cluster has two low-lying isomers with an energy difference of 0.03 eV, which can be viewed as five Si atoms capping a pentagon face of the Ti@Si14 motif and surrounding the surface Ti atom. The Ti2Si20− cluster has three low-lying isomers within an energy range of 0.02 eV. They all have fused cages encapsulating the two Ti atoms. More specifically, the 20–I and 20–III isomers present two identical 13-vertex cages sharing six Si atoms with C2 symmetry. The 20–II isomer has the Ti@Si14 motif and a 10-vertex cage fused by sharing a Ti atom and three Si atoms.
3.2 Photoelectron Spectra of TimSi −n
The experimental PES of TimSi −n (m = 1‒2, n = 14‒20) clusters recorded with 248 nm photons are given in Fig. 2 (blue lines), compared with the computed spectra (black lines). As the anions are open-shell with a single unpaired electron, both singlet and triplet final states have been taken into consideration in the simulated PES. The VDE values, representing the vertical binding energy of an excess electron to the corresponding neutral cluster, are plotted in Fig. 3.

Photoelectron spectra of low-lying isomers of TimSi −n (m = 1‒2, n = 14‒20) from experiment (blue lines) and theory (black lines). In the simulations, Gaussian broadening with a width of 0.06 eV is used

VDEs of TimSi −n (m = 1‒2, n = 14‒20) clusters as a function of n. Red dots: experimental values with error bars; black squares: calculated results for m = 1; black triangles: calculated results for m = 2, in which the calculated VDEs of Ti2Si14− and Ti2Si15− come from their third and second isomer, respectively
For Ti1Si14−, the experimental spectrum has one characteristic peak corresponding to the ground state transition X with VDE = 4.31 eV (Fig. 2a). The simulated PES of the 14–I isomer shows one major peak centered at 4.26 eV, while the 14–II isomer shows multiple peaks, significantly deviating from the experimental spectrum. Thus, the 14–I isomer is the most probable structure detected in the experiment. For Ti1Si15−, Ti1Si16− and Ti1Si17−, the experimental spectrum presents a low-intensity X band with VDE values of 3.66, 2.35 and 3.15 eV, and a prominent A band centered at 4.32, 4.48 and 4.20 eV, respectively. Particularly, Ti1Si16− shows a large gap of 1.97 eV between the X band and A band, which is the HOMO–LUMO gap of the neutral Ti1Si16 cluster with X and A bands corresponding to photodetachment of an electron from the singly occupied molecular orbital (SOMO) of the Ti1Si16− anion and from the HOMO of the neutral Ti1Si16 species, respectively. The simulated PES of 15–I, 16–II and 17–I isomers have a prominent peak at 4.38, 4.49 and 4.22 eV, and a minor peak at 3.59, 2.32 and 3.14 eV, respectively, in good agreement with the experimental data for each size. In contrast, the 16–I isomer has a lower VDE (2.20 eV), and the 15–II and 17–II isomers exhibit multiple peaks in the A band; hence they can be excluded from the clusters obtained in the laboratory. For Ti1Si18− and Ti1Si19−, the experimental spectrum has a low-intensity X band at 3.27 and 3.49 eV, respectively, followed by multiple peaks above 3.90 eV corresponding to A and B bands. The computed PES well reproduce the features of the experimental spectra, showing X bands at 3.24 and 3.48 eV for Ti1Si18− and Ti1Si19−, and prominent A bands with several overlapping peaks, respectively. Therefore, for the mono-Ti-doped anionic Si clusters, the lowest-energy isomers found by CGA are usually the structures obtained in the experiment, with the exception of Ti1Si16−, for which the higher energy isomer (0.06 eV above the lowest-energy one) is the more probable cluster species detected in the experiment.
For bi-doped Si clusters, the density of electronic states near the Fermi level is more complicated than that for the mono-doped systems, giving broadened features in the PES data and making definitive identification more difficult. For Ti2Si14− and Ti2Si15−, the experimental spectrum reveals a low-intensity X (Xʹ) peak at 3.04 and 2.98 (3.25) eV, and an A band at 3.52 and 3.52 eV, respectively, followed by a broad high-intensity peak (here assigned as B band) centered at about 4.26 eV (Fig. 2b). The simulated PES of three isomers of Ti2Si14− display a first peak with VDE of 3.27, 3.17 and 3.05 eV, respectively. Among them, the third one shows good accordance with the experimental PES, while the simulated PES of the first two isomers of Ti2Si14− deviate from the experimental observation. The simulated PES of 15–I exhibits multiple peaks with the first and second peaks at 3.31 and 3.74 eV, in good accordance with the experimental Xʹ and A bands, respectively. The 15–II isomer has two low-intensity peaks at 3.17 and 3.35 eV, followed by a broad peak above 4.0 eV, coinciding with the experimental X, Xʹ and B bands, respectively. Therefore, we speculate that these two isomers with their small energy difference (only 0.01 eV at 0 K) may coexist in the experiment. Indeed, the calculated free energies of Ti2Si15−–I and Ti2Si15−–II isomers (with inclusion of zero-point energy and vibrational entropy effect) would lead to a crossover at a temperature around 80 K.
For Ti2Si16−, the experimental spectrum reveals X and A peaks at 3.40 and 3.90 eV, respectively, followed by a broad high-intensity peak centered at about 4.25 eV. The simulated PES of two isomers of Ti2Si16− display a first peak with VDE of 3.44 and 3.09 eV, respectively. Among them, the 16–I shows good accordance with the experimental PES, while the 16–II deviates from the experimental observation. For Ti2Si17− and Ti2Si18−, the experimental spectrum shows a low-intensity and barely resolved X band at 3.32 and 3.57 eV, overlapping with an A band at 3.58 and 3.76 eV, respectively, along with broad high-intensity peaks above 4.1 eV. The simulated spectrum of 17–I (18–I) isomer displays X and A peaks at 3.32 (3.57) and 3.66 (3.78) eV, respectively, followed by higher peaks in the energy region above 4.1 eV, in agreement with the experimental observation. Therefore, the 17–I and 18–I isomers are probable structures detected in the experiment. The 17–II and 18–II isomers, on the other hand, have higher energies by 0.20 and 0.17 eV than the ground states, respectively, and their simulated spectra are distinct from the experimental ones.
3.3 Bonding and electronic properties of TimSi −n
To better understand the bonding nature of the TimSi −n clusters, we examined the natural charge, bond length, bond order, and binding energies of the ground state configurations which have been identified by the experimental and computed PES. As revealed in Table 2, for the mono-doped Si clusters, the endohedral Ti atom gains 2.09‒3.28 e from the Si atoms. Generally speaking, the amount of electrons transferred to the Ti atom decreases as the cluster grows bigger due to the increasing Ti–Si bond length (from 2.73 to 2.89 Å); accordingly, the Ti–Si bond order reduces from 0.62 to 0.52, as displayed in Fig. 4a. On the contrary, the Si–Si bond length decreases from 2.54 to 2.45 Å and the bond order increases from 0.44 to 0.71 as the cluster size increases, showing a mixed covalent-metallic bond nature.

Average Ti–Si, Si–Si, and Ti–Ti bond lengths (BL), Wiberg bond orders (BO), binding energies (Eb) and HOMO–LUMO gap (EHL) for the ground state structures of TimSi −n (m = 1‒2, n = 14‒20) cluster as a function of the number of Si atoms. The left column and right column correspond to m = 1, and m = 2, respectively. The purple squares, red dot, and cyan triangles correspond to the Ti–Si, Si–Si, and Ti–Ti bond, respectively
For the bi-doped Si clusters, the central Ti atom gains 0.79‒2.98 e from the Si atoms, while a smaller amount of electrons transfers to the Ti atom on the cluster surface. As n increases and the emerging sub-cage become larger, the natural charge decreases for the central Ti atom and increases for the surface Ti atom. For Ti2Si20− with its fused-cage configuration, the two Ti atoms are equivalent and carry almost identical charges (~ 2.0 e). The average Ti–Si bond length varies little (2.72‒2.76 Å), whereas the Si–Si bond length decreases from 2.54 to 2.40 Å and the bond order increases from 0.47 to 0.77 as the cluster size increases, due to the transformation of the cage structure to the Ti@Si14-based structure. In cage structures, the Ti–Ti bond lengths are 2.55‒2.71 Å, very close to the Si–Si bond lengths, which implies that the exposed Ti atoms play the role of Si atoms. With the excess Si atoms attaching to the surface Ti atom, the Ti–Ti bonds lengths are elongated to 3.08‒3.17 Å; when the two Ti atoms are encapsulated by fused cages at size 20, the Ti–Ti bond length is 2.68 Å. The Ti–Ti bond lengths in cage-like structures are much longer than the measured Ti–Ti bond length of 1.94 Å in a Ti dimer, and shorter than the nearest neighboring distance of 2.89 Å in the bulk Ti solid [60, 61], while Ti–Ti bond lengths in the Ti@Si14-based structures are substantially longer than both two values. This suggests a stronger interaction between the two Ti atoms in cage-like structures than that in Ti@Si14-based structures. The Wiberg bond order of Ti–Ti bonds is 0.79‒0.88 in cage-like structures (n = 14‒15, 20) and 0.43‒0.60 in Ti@Si14-based structures (n = 16‒19), respectively, in line with the trend of the Ti–Ti bond lengths.
The binding energy of Ti1Si −n clusters increases with size for n = 14‒16 (5.09‒5.13 eV/atom), decreases again at n = 17 (5.11 eV/atom), then rises slowly with size for n = 18‒20 (5.12‒5.13 eV/atom). The local minimum (n = 17) corresponds to the formation of the Ti@Si14 motif. The local maxima found at Ti1Si15− and Ti1Si16− designate their high stabilities, which is consistent with the experimental mass spectra showing abundant Ti1Si15− and Ti1Si16− [27]. The Ti1Si17− cluster with hanging Si buds has lower binding energy, and the binding energy increases with n as the Si bud evolves into a sub-cage. For the Ti2Si −n clusters, the binding energy decreases as the cluster grows bigger for n = 14‒18 (5.19‒5.17 eV/atom) due to the increasing number of attached Si atoms. For n = 19‒20, the attached Si atoms start to evolve into a sub-cage, and hence the binding energy rises slightly with n, i.e., 5.18‒5.19 eV/atom.
The HOMO–LUMO gap of the Ti1Si −n clusters reduces as cluster size increases from 2.50 to 0.75 eV. As an exception, the Ti1Si16− anion has a small HOMO–LUMO gap of 0.75 eV ascribed to the occupancy of SOMO. Generally speaking, the smaller-sized Ti1Si −n clusters (n = 14‒15) with cage-like structures possess larger HOMO–LUMO gaps than the larger size ones (n = 17‒20) with Ti@Si14-based structures. The Ti2Si −n clusters have smaller HOMO–LUMO gap than that of Ti1Si −n . As the cluster size increases, the gap decreases for n = 14‒17 from 1.38 to 0.95 eV, and then increases with n due to the formation of fused cages.
3.4 Ti1Si16 and Ti2Si15 as superatoms
The minimum VDE values appeared at Ti1Si16− and Ti2Si15− for m = 1 and m = 2, respectively (shown in Fig. 3), which inspire us to study if the Ti2Si15− anion has an electron occupying on the SOMO and the corresponding neutral one has a closed electronic shell structure similar to the neutral Ti1Si16 cluster. To confirm this hypothesis and gain further insights, we examine the electronic structures of the neutral Ti1Si16 and Ti2Si15 clusters, which are acquired by fully optimizing the structures of various isomers of Ti1Si16− and Ti2Si15− anions. The obtained neutral clusters have almost identical structures as their anionic counterparts, except that the outer Ti atom exchanges with an apical Si atom in the distorted FK isomer (Ti2Si15–III). The FK and distorted FK isomers of neutral Ti1Si16 (Ti1Si16–I and Ti1Si16–II), as well as the FK and distorted FK isomers of neutral Ti2Si15 (Ti2Si15–II and Ti2Si15–III) are nearly degenerate (energy difference less than 0.07 eV, see Table S1 of Supporting Information). The Ti@Si14-based structure of Ti2Si15 (Ti2Si15–I) is energetically less favorable than the FK isomer by 0.21 eV.
The energy levels and spatial distributions of the occupied molecular orbitals of these two neutral clusters (including ground state configuration and metastable isomers) are illustrated in Fig. 5 and Fig. S4 of Supporting Information. Both neutral Ti1Si16− and Ti2Si15 clusters in their ground state behave as superatoms with orbitals can be clearly categorized into superatomic orbitals, leading to a closed electronic shell configuration (1S)2(1P)6(1D)10(1F)14(2S)2(2P)6(1G)18(2D)10. Obviously, the HOMO of the Ti1Si16 cluster has 2D character, while its LUMO corresponds to a 1H orbital. Similar behavior is found for the bi-doped Si cluster as shown in Fig. 5b. Although the orbital energy sequences may be slightly different from each other, their overall electronic configurations are the same. The calculated HOMO–LUMO gaps are 2.98 eV for Ti1Si16 and 1.23 eV for Ti2Si15, respectively, both suggesting rather high chemical stability. Despite of its relatively smaller HOMO–LUMO gap, the discovery of Ti2Si15 superatom suggests that both Ti atoms contribute four electrons for electron counting of electronic shell.

Energy levels and superatom orbitals for the FK isomers of a Ti1Si16 and b Ti2Si15. The number of horizontal line corresponds to the degeneracy of energy level. The HOMO–LUMO gaps are labeled on the corresponding level in the unit of eV. The unoccupied orbitals are indicated in blue
4 Conclusions
A systematic investigation of the structural and electronic properties of anionic TimSi −n (m = 1‒2, n = 14‒20) clusters have been performed using DFT calculations at the HSE06/aug-cc-pVDZ level of theory, combined with experimental photoelectron spectroscopy. The ground states are identified by comparing the experimental and simulated photoelectron spectra. For Ti1Si −n clusters, the smaller sized clusters (n = 14‒16) have cage structure with Ti atoms embedded in the center, while larger size clusters (n = 17‒20) adopt the Ti@Si14 motif capped by apical Si buds. For Ti2Si −n clusters, the smaller sized clusters (n = 14‒15) have cage structure with one Ti atoms encapsulated in the center and the other Ti atom staying on the cluster surface; while for n = 16‒20, the Ti@Si14-based structure starts to dominate the low-lying isomers and the excess Si atoms start to evolve into a sub-cage enclosing the Ti atom on the cluster surface. Natural population analysis reveal that the Ti atoms possess negative charges acting as electron acceptors in all clusters, while less amount of electron is transferred to the surface Ti atom. The neutral Ti1Si16 and Ti2Si15 clusters are identified as superatoms with closed electronic shells of (1S)2(1P)6(1D)10(1F)14(2S)2(2P)6(1G)18(2D)10. Except for the Ti1Si16 cluster, the newly found Ti2Si15 cluster with closed electronic shells and high stability may also suitable as building blocks for novel nanomaterials.
References
J.J. Zhao, Q.Y. Du, S. Zhou, V. Kumar, Chem. Rev. (2020). https://doi.org/10.1021/acs.chemrev.9b00651
M.F. Jarrold, Science 252, 1085 (1991)
S.N. Khanna, P. Jena, Phys. Rev. B 51(19), 13705 (1995)
V. Kumar, Y. Kawazoe, Phys. Rev. Lett. 87(4), 045503 (2001)
S.N. Khanna, B.K. Rao, P. Jena, Phys. Rev. Lett. 89(1), 016803 (2002)
V. Kumar, T.M. Briere, Y. Kawazoe, Phys. Rev. B 68(6), 155412 (2003)
U. Röthlisberger, W. Andreoni, M. Parrinello, Phys. Rev. Lett. 72(5), 665 (1994)
C.L. ReisJ, M. Pacheco, J. Phys. Condens. Matter 22(3), 035501 (2010)
C.L. Reis, J.M. Pacheco, Phys. Rev. B 82(15), 155440 (2010)
J. Wang, Q.M. Ma, R.P. Xu, Y. Liu, Y.C. Li, Phys. Lett. A 373(32), 2869 (2009)
H. Tsunoyama, H. Akatsuka, M. Shibuta, T. Iwasa, Y. Mizuhata, N. Tokitoh, A. Nakajima, J. Phys. Chem. C 121, 20507 (2017)
K. Koyasu, M. Akutsu, M. Mitsui, A. Nakajima, J. Am. Chem. Soc. 127(14), 4998 (2005)
X. Huang, S.J. Lu, X. Liang, Y. Su, L. Sai, Z.G. Zhang, J. Zhao, H.G. Xu, W. Zheng, J. Phys. Chem. C 119(20), 10987 (2015)
S. Neukermans, X. Wang, N. Veldeman, E. Janssens, R.E. Silverans, P. Lievens, Int. J. Mass Spectrom. 252(2), 145 (2006)
K. Koyasu, J. Atobe, M. Akutsu, M. Mitsui, A. Nakajima, J. Phys. Chem. A 111(1), 42 (2007)
H. Kawamura, V. Kumar, Y. Kawazoe, Phys. Rev. B 71(7), 075423 (2005)
L.J. Guo, X. Liu, G.F. Zhao, Y.H. Luo, J. Chem. Phys. 126(23), 234704 (2007)
A. Willand, M. Gramzow, S.A. Ghasemi, L. Genovese, T. Deutsch, K. Reuter, S. Goedecker, Phys. Rev. B 81(20), 201405 (2010)
N.A. Borshch, N.S. Pereslavtseva, S.I. Kurganskii, Russ. J. Phys. Chem. 88(10), 1512 (2014)
C. Dong, L. Han, J. Yang, L. Cheng, Int. J. Quantum Chem. 119(18), e25978 (2019)
D. Bandyopadhyay, J. Appl. Phys. 104(8), 084308 (2008)
D. Bandyopadhyay, M. Kumar, Chem. Phys. 353(1), 170 (2008)
W. Pei, S. Zhou, Y. Bai, J. Clust. Sci. 31, 627 (2020)
P. Sen, L. Mitas, Phys. Rev. B 68(68), 155404 (2003)
T. Iwasa, A. Nakajima, J. Phys. Chem. C 116(26), 14071 (2012)
M.I.A. Oliveira, R. Rivelino, F.D.B. Mota, G.K. Gueorguiev, J. Phys. Chem. C 118(10), 5501 (2014)
M. Ohara, K. Koyasu, A. Nakajima, K. Kaya, Chem. Phys. Lett. 371(3), 490 (2003)
S. Furuse, K. Koyasu, J. Atobe, A. Nakajima, J. Chem. Phys. 129(6), 064311 (2008)
K. Koyasu, J. Atobe, S. Furuse, A. Nakajima, J. Chem. Phys. 129(21), 214301 (2008)
M.B. Torres, E.M. Fernández, L.C. Balbás, J. Comput. Methods Sci. Eng. 7(3,4), 241 (2007)
J.U. RevelesS, N. Khanna, Phys. Rev. B 74(3), 035435 (2006)
J. He, K. Wu, C. Liu, R. Sa, Chem. Phys. Lett. 483(1–3), 30 (2009)
M.B. Torres, E.M. Fernández, L.C. Balbás, Phys. Rev. B 75(75), 205425 (2007)
L.J. Guo, G.F. Zhao, Y.Z. Gu, X. Liu, Z. Zeng, Phys. Rev. B 77(77), 195417 (2008)
J. Zhao, R. Shi, L. Sai, X. Huang, Y. Su, Mol. Simul. 42(10), 809 (2016)
B. Delley, J. Chem. Phys. 113(18), 7756 (2000)
L.W. Sai, X. Wu, X.M. Huang, J.J. Zhao, J. Clust. Sci. 28(3), 1729 (2017)
X. Wu, X. Liang, Q. Du, J. Zhao, M. Chen, M. Lin, J. Wang, G. Yin, L. Ma, R.B. King, B.V. Issendorff, J. Phys. Condens. Matter 30, 354002 (2018)
L.W. Sai, X. Wu, N. Gao, J.J. Zhao, R.B. King, Nanoscale 9, 13905 (2017)
X. Wu, L. Sai, S. Zhou, P. Zhou, M. Chen, M. Springborg, J. Zhao, Phys. Chem. Chem. Phys. 22, 12959 (2020)
X. Huang, H.G. Xu, S. Lu, Y. Su, R.B. King, J. Zhao, W. Zheng, Nanoscale 6(24), 14617 (2014)
X. Wu, S.-J. Lu, X. Liang, X. Huang, Y. Qin, M. Chen, J. Zhao, H.-G. Xu, R.B. King, W. Zheng, J. Chem. Phys. 146(7), 044306 (2017)
X. Wu, S. Zhou, X. Huang, M. Chen, R.B. King, J. Zhao, J. Comput. Chem. 39, 2268 (2018)
X.Q. Liang, X.J. Deng, S.J. Lu, X. Huang, J. Zhao, H.G. Xu, W.J. Zheng, X.C. Zeng, J. Phys. Chem. C 121, 7037 (2017)
X. Liang, X. Kong, S.-J. Lu, Y. Huang, J. Zhao, H.-G. Xu, W. Zheng, X.C. Zeng, J. Phys. Condens. Matter 30, 335501 (2018)
Y. Wang, X. Wu, J. Zhao, J. Clust. Sci. 29, 847 (2018)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77(18), 3865 (1996)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 78, 1396 (1997)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, Gaussian 09, Revision A.1 (Gaussian Inc., Wallingford, 2009)
T.H. Dunning Jr, J. Chem. Phys. 90(2), 1007 (1989)
A.V. Krukau, O.A. Vydrov, A.F. Izmaylov, G.E. Scuseria, J. Chem. Phys. 125(21), 224106 (2006)
C. Adamo, J. Chem. Phys. 110(13), 6158 (1999)
A.E. Reed, R.B. Weinstock, F. Weinhold, J. Chem. Phys. 83(2), 735 (1985)
A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88(6), 899 (1988)
J.E. Carpenter, F. Weinhold, J. Mol. Struct. Theochem 169(4), 41 (1988)
D.J. TozerN, C. Handy, J. Chem. Phys. 109(23), 10180 (1998)
J. Akola, M. Manninen, H. HäKkinen, U. Landman, X. Li, L.S. Wang, Phys. Rev. B 62(19), 13216 (2000)
L. Ma, B.V. Issendorff, A. Aguado, J. Chem. Phys. 132(10), 104303 (2010)
F.C. FrankJ, S. Kasper, Acta Cryst. 11(3), 184 (1958)
R.W.G. Wyckoff, Crystal Structures (Interscience Publishers, New York, 1963)
M. Doverstal, B. Lindgren, U. Sassenberg, C.A. Arrington, M.D. Morse, J. Chem. Phys. 97(10), 7087 (1992)
Acknowledgements
We would like to express thanks to Prof. Vijay Kumar for stimulating discussion. This work was supported by the National Natural Science Foundation of China (91961204, 11974068 and 11804076), the Key Project of Natural Science Foundation of Tianjin City (No. 17JCZDJC30100), the Fundamental Research Funds for the Central Universities of China (DUT20LAB110 and DUT20LAB203), and the Supercomputing Center of Dalian University of Technology.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, X., Du, Q., Zhou, S. et al. Structures, stabilities and electronic properties of TimSi −n (m = 1‒2, n = 14‒20) clusters: a combined ab initio and experimental study. Eur. Phys. J. Plus 135, 734 (2020). https://doi.org/10.1140/epjp/s13360-020-00745-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjp/s13360-020-00745-6