
Abstract
Magnesium oxide (MgO) is a versatile metal oxide with wide applications in electrical, chemical and pharmaceutical industries. Preparation of nano-MgO helps to enhance key physicochemical properties for optimal performance in industrial applications. In the present work, MgO nanoparticles were synthesized by magnesium nitrate precursor in ethanol. Different gelling agents were employed to investigate potential variations in the crystallinity and thermal behavior of the sol–gel product to control the nanoparticle’s size distribution. The crystal and thermal characteristics of the synthesized MgO nanoparticles were studied using X-ray diffraction (XRD) and Thermogravimetry-Differential Scanning Calorimetry (TG-DSC) analysis. The XRD data showed that all the samples demonstrated crystallinity except sample B which was amorphous. The TG-DSC characterization showed a three stage thermal decomposition for all the samples, leading to the formation of MgO nanoparticles. Based on the thermal analysis data, different calcination temperatures were selected to investigate their effects on stability, and the sample prepared with tartaric acid as a gelling agent and calcined at 500oC demonstrated the smallest average particle size of 58.7 nm obtained from dynamic light scattering (DLS) analysis. Further, this sample was subjected to XRD, FTIR and TEM analysis which reveals that the calcination yielded impure, 30 nm sized spherical shaped, agglomerated MgO nanoparticles. Additionally, the physicochemical characteristics of the selected sample reveals that pure MgO nanoparticles with uniform morphology can be obtained via alteration of calcination time and heating rate.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 INTRODUCTION
Sol–gel synthesis is classified as a wet-chemical technique used to synthesize inorganic and organic-inorganic hybrid nanomaterials [1]. The term ‘sol’ represents the dispersion of colloidal particles in a liquid [2], and “gel” depicts the interconnected rigid network of polymeric chains. Sol–gel process mechanism yields polymeric gel in three major steps; gelation, hydrolysis and poly-condensation [3]. The polymeric gel generated from the sol–gel process further undergoes downstream process including aging, drying and calcination to form nanoparticles. Amongst various nanoparticle synthesis process, sol–gel is recognized as one of the most common methods for the synthesis of metal oxide nanoparticles [4]. It is also a suitable method for metal oxide fabrication into composites, porous materials and coatings with high chemical stability and mechanical strength [5]. Metal oxide nanoparticles is one of the most diverse class of materials with unique characteristics and enhanced chemical and physical properties [6]. These oxide nanoparticles are used in emerging technologies such as gas sensing, energy storage, catalysis [7] and biomedical applications [8].
Amongst all metal oxide nanoparticles, Magnesium oxide (MgO) nanoparticles have demonstrated potential for enhanced electrical, thermodynamic, chemical [9] and pharmaceutical applications [10]. It has been used as a catalyst [11, 12], refractory materials [13], superconducting materials, and for hazardous waste treatment [14]. It has also been used in biological applications such as antibacterial agents [15], for heart burn treatment, bone regeneration [16] and cancer therapy [17–19]. The morphology of MgO nanoparticles is an important property that affects the functional properties of MgO nanoparticles, and this has led to the synthesis of different morphologies of MgO nanoparticles with unique properties and functionalities [20–22]. Hexagon shaped MgO nanoparticles with size 50–100 nm have previously been synthesized through hydrothermal methods [23]. Similarly, porous MgO nanoparticles as small as 4.1 nm and irregular shaped MgO nanoparticles of about 9 nm were synthesized using microwave-induced combustion and solvent mixed spray pyrolysis technique respectively [24, 25]. Hence, the present study is an attempt to synthesize morphologically different MgO nanoparticles using different gelling agents and calcination temperatures as characteristic process variables in sol–gel synthesis. It has previously been demonstrated that sol–gel synthesis of MgO nanoparticles facilitates the generation of small size nanoparticles compared to other chemical synthesis methods [26–30]. The properties of the gelling agent is highly essential for the formation of mechanically stable gels in sol–gel synthesis as it facilitates the development of robust inter-particulate linkages [31]. Citric acid [32], tartaric acid [33], oxalic [34] and glycolic acid [35] have previously been employed as gelling agents in sol–gel synthesis, and most of these gelling agents demonstrated homogeneous sol–gel products. However, these studies were carried out to synthesize metal oxides using metal nitrates as precursors [36]. The extent of the influence of various gelling agents on the physicochemical characteristics of sol–gel product is not well established. These difference could result from the molecular structure and solubilization characteristics of the gelling agent during nanoparticle formation. Thus, the aim of the present study is to synthesize MgO nanoparticles using sol–gel method with different gelling agents and varying calcination temperature in order to probe the nanoparticle formation process and to characterize the physicochemical features of the sol–gel products by Thermogravimetry and Differential Scanning Calorimetry (TG-DSC), X-ray Diffraction (XRD), and Dynamic Light Scattering (DLS). Insights into the chemical reactions driving the formation of sol–gel product and the thermal degradation characteristics for the formation of MgO nanoparticles are discussed. Moreover, the calcinated MgO nanoparticle sample, below 100 nm, was subjected to XRD, Fourier Transform–Infrared (FTIR) spectroscopy and Transmission electron microscopy (TEM) analysis to study their physicochemical characteristics. These characteristics leaves a valuable clue to engineer the morphology, the size and the crystalline phase of MgO nanoparticle via calcination temperature and time alteration.
2 EXPERIMENTAL PROCEDURE
2.1 Materials Used
Magnesium nitrate hexahydrate (98%) was obtained from Alfa Aesar (USA). Oxalic acid (99.5%), tartaric acid (99.5%), and citric acid (99.9%) were obtained from Merck (USA), Acros organics (Belgium) and Fisher scientific (Singapore) respectively. Ethanol (99.4%) was also obtained from Fisher scientific (Singapore). All these chemicals were directly used in the synthesis process with no initial pre-treatment.
2.2 Synthesis of MgO Nanoparticles
Equimolar concentrations of precursor and gelling agent were individually dissolved in ethanol before mixing to form a gel matrix with pH 5. After the formation of 3D network of magnesium gel, the matrix was allowed to age for 12 h and dry at 100°C for 24 h to form the sol–gel powder [37–39] as shown in Fig. 1. The sample notations for sol–gel products obtained from magnesium nitrate with different gelling agents are shown in Table 1. The synthesized sol–gel powder was then calcined at three different temperatures for 2 h at a heating rate of 5°C/min in a box furnace to form pure MgO nanoparticles. In the present work, samples A, B and C, developed from magnesium nitrate precursor and gelling agents oxalic acid, tartaric acid and citric acid in a 1 : 1 ratio respectively, were used for the synthesis of MgO nanoparticles. For sample D, a mixture of all three gelling agents was added to magnesium nitrate precursor in 3 : 1 : 1 : 1 ratio to form MgO nanoparticles.

A flowchart for sol–gel synthesis of MgO nanoparticles.
2.3 Characterization of MgO Nanoparticles
The range of calcination temperature was determined from thermal degradation and the heat flow pattern of the samples was obtained through Thermo Gravimetry–Differential Thermal Analysis (Thermal Gravimetric Analyzer, Mettler Toledo,Switzerland). 20 mg of sample was heated from 30–1000°C at a heating rate of 20°C/min under an inert condition for thermal analysis. Calcination of the sol–gel product was performed in a box furnace (Carbolite 1200°C heavy duty box furnace, Cole-Parmer,USA) for 2 h. The average particle size of MgO nanoparticles was determined using Dynamic Light scattering technique (DLS) (Malvern® Zeta sizer nano series, United Kingdom). The crystallinity of the sol–gel products was analyzed using X-Ray Diffractometer (Bruker® Advanced D8 XRD, USA). The powder samples were pressed, evenly distributed in the sample holder and placed at an angle of 45° for XRD analysis. The intensity of the beam was set to 40 kV in order to avoid noise peaks. The selected sample with average particle size below 100 nm was subjected to XRD, Fourier Transform Infrared (Bruker Vector 22 FTIR) and Transmission electron microscope (Hitachi HT-7700 TEM) analysis to study their crystallinity, functional group and morphology respectively.
3 RESULTS AND DISCUSSION
The dissolution of the precursor and gelling agent in ethanol immediately resulted in a white color gel-like precipitate, and the system was stirred for 2 h to complete the gelation process. The obtained gel was allowed to age for 12 h at room temperature and dried in an oven for 24 h at 100°C before the formation of MgO nanoparticles. After the drying process, a pale yellow sol–gel powder was obtained in all the samples (A, B, C and D) as shown in Fig. 2.

Nature of gel before and after aging and drying during sol–gel synthesis of MgO nanoparticles.
3.1 Crystalline Characteristics of the Sol–Gel Product
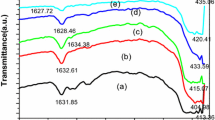
The crystal formation in the sol–gel powders were studied using X-ray diffraction analysis. The XRD patterns of the sol–gel products from samples A, B, C and D are shown in Fig. 3. The presence of a characteristic crystal peak is observed for samples A, C and D as shown in Figs. 3a, 3c, 3d. The presence of additional peaks and peak shifts (marked with the symbol *) is also observed in the crystalline samples, and this is due to the presence of impurities, thermally degraded molecular moieties during drying process and/or unreacted molecules associated with the precursor and gelling agent. The samples were JCPDS (Joint Committee on Powder Diffraction Standards) analyzed to identify phase formation activity and crystallinity from standard peaks. As no characteristic XRD peak for crystals was observed for sample B (Fig. 3b), it is categorized as amorphous powder. The reason for amorphous formation is that the drying temperature or time is not sufficient to convert the sol–gel product into crystalline. This can also be attributed to the lack of binding ability between the magnesium precursor and gelling agent–tartaric acid molecules due to insufficient drying temperature and time [40, 41]. XRD peaks of sample A (Fig. 3a) and C (Fig. 3c) matched with JCPDS files–PDF 26-1223 and PDF 14-101 corresponding to magnesium oxalate dihydrate [37] and nitromagnesite [42] respectively. No JCPDS files matched with the peaks observed for sample D (Fig. 3d), hence the data was analyzed with COD (Crystallography Open Database) file and showed a complex crystal (C16H34MgN4O14) with COD no: 96-151-9079 [43] along with impurity peaks and peak shifts. The crystals formed as sol–gel product for each sample along with their reference JCPDS/COD file numbers are summarized in Table 2.

XRD patterns of samples A (a), B (b), C (c) and D (d).
3.2 Proposed Equation for the Formation of Sol–Gel Product
The predicted chemical structures of sol–gel products formed in all four samples A, B, C and D are shown in the Fig. 4. These predictions are made based on the data obtained from X-ray diffraction analysis (Fig. 3). The predicted structures represent gel products formed immediately after mixing the precursor (magnesium nitrate) and gelling agents dissolved in the solvent (ethanol). The chemical structure of magnesium oxalate dihydrate formed in sample A is referenced in reaction scheme 1 (Equation-1). The ethanol solvent is not involved in the reaction as it only acts as a solvent to dissolve the precursor and gelling agents in sample A, B and C. The unreacted ethanol evaporates during aging and drying as the boiling point of ethanol is about 78°C. However, ethanol is anticipated to be involved in the product formation reaction of sample D (Equation-4) since multiple gelling agents are dissolved in ethanol. It was noted that a two-step process is involved in the formation of magnesium oxalate dihydrate in sample A. When the ethanol dissolved magnesium nitrate hexahydrate and oxalic acid were mixed together, nitrite ion from magnesium nitrate and the alcohol molecule from oxalic acid are dislodged from their native molecular sites due to constant stirring and pH change. The initial pH of ethanol dissolved precursor and oxalic acid were neutral. When both solutions are mixed together through constant stirring, a sudden shift of pH to pH 5 is observed due to protonation, leading to the dislodgment of alcohol and nitrite from their respective native molecular sites. Tamás et al. (2006), also explained that pH change promotes the dissociation of ions and helps in ion exchange [44]. Carbon-carbon chains from unstable oxalic acid molecules binds onto unstable magnesium to form stable magnesium oxalate dihydrate molecules. Water molecule, nitrite and alcohol generated from the process are evaporated or volatilized during aging and drying process or become part of the impurities as mentioned in the XRD data (Fig. 3A).

Chemical reaction scheme for sol–gel synthesis and products generated for sample A, B, C and D.
The sol–gel product formed in sample B was found to be an amorphous solid from XRD analysis (Fig. 3B). The formation of amorphous solid may be due to the presence of weak nucleophilic sites on reactive groups, further allowing random arrangements of atoms after drying at 100°C. However, drying temperature higher than 100°C may results in a crystalline material generated from sample B [45]. Since, amorphous solids do not possess stable structures, as atoms are randomly arranged [46], it is a challenge to propose chemical structures for such products. Hence, the structure proposed in Fig. 4 (Equation-2) is an unstable structure predicted to have an altering molecular structure until transformation into powder form after aging and drying. Likewise, the formation of nitromagnesite in sample C as shown in Equation-3 was entirely due to the magnesium nitrate precursor. The gelling agent (citric acid) and the solvent (ethanol) facilitated ethanol-dissolved magnesium nitrate to transform into a nitromagnesite crystal. The ethanol may evaporate due to aging and drying and the ethanol-dissolved citric acid may remain as an impurity. In sample D, a complex crystal was formed as a result of reaction between three gelling agents and a nitro-magnesium precursor as predicted from XRD analysis. Along with the complex mixture described Equation-4, the alcohol and oxygen groups released from the end carbon chain of the gelling agents and ethanol may present in the product system. These molecular groups may be evaporated while aging and drying or may remain as impurities. It can be noted that the compounds formed during gel formation are predicted to have nucleophilic sites. These nucleophilic sites are responsible for networking with neighboring molecules to form rigid gels [47].
3.3 Thermal Analysis of Sol–Gel Products
Thermal analysis of synthesized sol–gel products were conducted using TGA to characterize sample weight loss due to increase in temperature at a constant heating rate, and DSC to profile the heat flow pattern either exothermic or endothermic. By correlating sample weight loss and heat flow pattern, molecules that are thermally degraded from the sample can be identified. The thermal analysis data showing the weight loss and heat flow are shown in Figs. 5a, 5b, 5c, 5d. As shown in Table 3, all four samples demonstrated a three-stage thermal degradation process irrespective of their crystallinity characteristics. The first degradation phase for all the samples represented endothermic thermal decomposition of entrapped or crystallizing water by vaporization [37]. The second degradation phase is due to endothermic decarboxylation in sample A [37, 48, 49], exothermic melting transition of unstable magnesium complex into magnesium peroxide in sample B, endothermic removal of nitrogen dioxide in sample C [50] and exothermic degradation of nitrogen dioxide, carbon, hydrogen, nitrogen and oxidation in sample D [51, 52]. The third degradation phase in sample A, C and D is due to the melting transition of unstable magnesium complexes to form MgO. It can also be inferred from the presence of a peak in the third degradation phase the potential binding of oxygen to sample D during melting transition [53]. For sample B, the third degradation phase resulted in endothermic formation of MgO as the second degradation phase produced thermally unstable magnesium peroxides [54]. All four samples experienced ~52–67% weight loss from sol–gel product to form stable MgO. The presence of different precursor and gelling agents in the synthesis of sol–gel products have unique impacts on the thermal degradation pathway of the reaction kinetics and this explains the thermo-molecular differences in the degradation phases of the samples.

TG-DSC analysis of sample A (5a), sample B (5b), sample C (5c) and sample D (5d).

(Contd.)
From DSC analysis, the melting transition for the amorphous sample B with tartaric acid as gelling agent is observed to be lower compared to other samples (refer to Table 3). However, sample D, which also contains tartaric acid, did not significantly leads to a lower melting transition temperature as the concentration of tartaric acid was less compared to sample B. This makes the sol–gel product from sample D, a complex mixture, rather than an amorphous material. Thus, a higher temperature is required to transform it into stable MgO. This demonstrates that tartaric acid plays a major role as a gelling agent to reduce melting transition temperature in sol–gel reactions. Also, the degree of crystallinity is vital in the formation of stable MgO crystals at lower temperatures. High temperatures are required to transform crystalline sol–gel products into MgO crystal as the atoms in the crystals are uniformly arranged. In contrast, atoms are arranged randomly in amorphous powders hence a lower energy is sufficient to form MgO crystals. This shows that in sol–gel synthesis of nanoparticles, the type and composition of gelling agent play an important role in influencing the melting transition temperature.
3.4 Proposed Thermal Decomposition Reactions for MgO Nanoparticle Formation
The formation of thermally stable magnesium oxide from samples A, B, C and D is modelled in the form of thermal degradation reaction as shown in Table 4. For all the samples, the first stage of degradation was observed between 70–210°C, and this is due to the decomposition of crystallizing water via vaporization. Kumar et al. [37] reported an initial degradation phase of sol–gel product up to 280°C, resulting from the decomposition of crystallizing water from the crystals. Within this initial degradation phase, crystallinity and the number of water molecules play a significant role in determining the first degradation temperature range for each sample. In the second stage, molecules with boiling point around 165–320°C are decomposed. As a result of decarboxylation in sample A, carbon dioxide is released from unreacted excess CO2 compounds in sol–gel product at 165–220°C in stage 2 as mentioned in Equation 1(b). The release of carbon-dioxide from sample A was validated by comparing the temperature range with previous literatures [37, 38, 48]. Jeevanandam et al. (2015) proposed a similar thermal degradation equation for the preparation of magnesium peroxide nanoparticles and mentioned that release of carbon dioxide occurs between 155–206°C [38]. However, Kumar et al (2008) proposed a transformation of MgC2O4 to MgO at 416–564°C [37] due to decarboxylation in the second stage with no intermediate decarboxylation reaction as mentioned in the present study. Dureswamy et al (2005) gave an insight on decarboxylation process at 319–396°C in a complex molecule. They explained that high degradation temperatures could be due strong crystalline bonding between reacting molecules [48]. In the stage 3, unstable magnesium complex further decomposes and strips off carbon and oxygen units and undergo atmospheric oxidation to form magnesium peroxide as shown in Equation 1(c). This correlated with a significant weight loss from 440–560°C and a melting transition of 525°C. As magnesium peroxide is not stable at high temperatures [54], calcination of sol–gel product above the melting transition temperature yields MgO.
For all the other samples, different molecules are decomposed in stages 2 and 3, resulting in the presence of exothermic peaks from DSC. The degradation of the unstable complex molecule [C6H12O13N2] and oxidation in the amorphous sample B are potential reasons for the sudden exothermic peak at 200–230°C and the formation of magnesium peroxide [55]. As mentioned in Equation 2(c), MgO2 formed in the second stage was transformed into stable MgO from 250–450°C as magnesium peroxide has a lower thermal stability [54]. Similarly, nitromagnesite from sample C in stage 2, as shown in Equation 3(b), experienced an exothermic reaction from 220–320°C, and this was anticipated to be as a result of the decomposition of nitrogen dioxide [50]. Within the temperature range of 340–510°C, peroxynitrate is degraded from the remaining unstable compound Mg[NO4] and this is inferred from the melting transition temperature of 380°C as stated in Equation 3(c) [53]. This results in the formation of magnesium peroxide by gaining an oxygen atom from the atmosphere, and the calcination process yields MgO after thermal molecular stabilization. Likewise, a sudden exothermic peak was observed in sample D from 240–330°C in stage 2 due to the decomposition of nitrogen dioxide as well as carbon, hydrogen and nitrogen from the complex crystal structure [51, 52] as indicated in Equation 4(b). In stage 3, as shown in Equation 4(c), oxygen binds onto magnesium at 360–475°C to form magnesium peroxide as indicated by the melting transition at 470°C, and the calcination process results in the formation of MgO.
3.5 Calcination of Sol–Gel Product
Depending on the proposed thermal degradation reaction of the different sol–gel products, the calcination temperature is identified based on the melting transition temperature required for the formation of MgO nanoparticles. It has been reported that calcination of sol–gel product above its melting transition temperature could lead to the formation of nanoparticles with different morphology [56, 57]. Yu and Wang [57] demonstrated that increasing calcination temperature from 300–800°C for 2 h at a heating rate of 2°C/min resulted in the formation of titanium dioxide nanotubes with enhanced crystallization from spherical structure. Gibson et al. [58] also revealed that 2 h of calcination led to an unusual increase in the crystallite size i.e. the calcination of Ca-deficient apatite at 700, 710, 720 and 730°C for 2 h leads to the formation of 23, 22, 28 and 145 nm sized crystallites. Three different calcination temperatures were investigated for each sample and the process was performed for 2 h at a heating rate of 5°C/min as shown in Table 5. The calcination process thermally transformed unstable magnesium peroxide to MgO nanoparticles with different morphologies.
3.6 Particle Size Distribution Analysis
The calcined powder samples were dissolved in distilled water for particle size distribution analysis. The average particle sizes of all the calcined samples are shown in Table 5. The average size of the different samples are mostly above 100 nm, and this is based on spherical approximation of the DLS system. Among all the calcined samples, MgO nanoparticle from sample B calcined at 500°C showed the smallest average particle size of 58.77 nm. Figure 6 shows the trend of average particle size of the different samples calcined for 2 h at three different temperatures. The data shows no trend between the different samples. Increasing calcination temperature affects the rate of particle formation and distribution, however, the extent of impact is dependent on the composition of the sol–gel product. Initially, sol–gel powder from each sample is irregular in shape. The calcination temperature of the sol–gel product was fixed after 25–150°C of melting transition temperature and depending on the thermal stability. The first calcination temperature which generated pure MgO which was predicted to be spherical in shape. Depending on the quantity and stability of MgO2 formed during melting transition, which is based on the crystallinity of the sol–gel powder, the size of spherical MgO nanoparticles may differ in the first calcination temperature of each sample. Only in sample A, the increment in the size was gradual due to the transformation from spherical to hexagon and from hexagon to elongated hexagon. This may be due to the relaxation of atomic lattices due to thermal energy [59]. In sample B and C, the first calcination temperature possibly yielded larger spherical particles which were converted into smaller spherical particles in the second calcination temperature. The third calcination temperature resulted in a mixture of spherical and hexagon shaped particles, explaining the larger particle size DLS data observed. In sample D, initial calcination yielded larger particle and an increase in temperature created a mixture of spherical and hexagon shaped particles comparatively smaller than the particles produced by initial calcination. The third calcination temperature yielded elongated hexagon and distorted by the spherical approximation of DLS analysis.

Effect of calcination temperature on average particle sizes of sol–gel products for MgO nanoparticles syntheiss.
This supports that the type and composition of gelling agent used for the formation of the sol–gel product play an important role in understanding the relationship between the calcination temperature and the average size of the nanoparticles. To support this argument, Keshmiri et al. (2004), in their work on developing novel TiO2 particles, reported that gelation conditions and the viscosity of the gelling solution have a strong effect on determining the sintering temperature and final product [60]. Also, the graph (Fig. 6) gives insight into calcination duration and temperatures suitable for generating small size nanoparticles from sol–gel products.
4 CHARACTERIZATION OF SELECTED MgO SAMPLE
Based on the DLS analysis, the sample B which was calcinated at 500°C was selected for further characterization to confirm their crystallinity. MgO phase formation was analysed using XRD analysis, functional group analysis using Fourier Transform-Infra red (FTIR) spectroscopy and morphology analysis using Transmission Electron Microscopy (TEM).
4.1 Crystallinity of Selected MgO Sample
Figure 7 shows the crystallinity of the selected calcined sample. The peaks in the calcined sample from the XRD data were matched with the JCPDS files PDF 71-1176, PDF 30-794, PDF 76-1363, PDF 35-821 and PDF 44-1482 corresponding to two different phases of magnesium oxide, magnesium peroxide, magnesium and magnesium hydroxide respectively. Some additional phase peaks were also present in the XRD pattern, indicating the presence of some impurities in the crystalline content. The peaks represent the formation of a mixed MgO phases along with peak shifts, traces of other magnesium crystalline phases and impurity peaks. The reason for the mixture of crystallinity in the sample may be due to the insufficient calcination time at the particular calcination temperature for the amorphous sol–gel product. The peaks provide an insight on the calcination mediated transformation of amorphous solid into MgO. In Fig. 7, most of the peaks are related to two different crystalline phases of magnesium oxide (PDF 71-1176 and PDF 30-794). Moreover, peaks related to magnesium per oxide (MgO2), magneisum hydroxide (Mg (OH)2) and unoxidized magnesium were also present as a result of existence of these additional, unreated atoms in the interstial position of MgO crystal phase. This shows that the sol–gel product (sample B) calcinated at 500°C for 2 h unable to completely converted into a pure MgO crystal. The calcination process converted the sample into two different phases of MgO which is reflected from the XRD peaks. The presence of unoxidized magnesium represents the insufficiency of the low calcination temperature for complete reaction as well as shorter period to transform MgO2 and Mg (OH)2 into MgO were used. When the calcination temperature is prolonged for a longer time, it leads to the oxidation of the sample [61]. Thus, these polycrystalline MgO product can be converted into an pure MgO by the prolongation of calcination time at the current calcination temperature (500°C). The Debye Scherer’s formula (D = kλ/βcos θ) has been utilized to determine the crystallite size of the calcinated sample. The full width half maximum (FWHM) value for the calcinated sample was identified to be 0.23° which was obtained from the highest intensity peak and the crystallite size was determined to be 37.36 nm. Literature shows that the primary particles required for the nanoparticle formation are crystallites [62]. Hence, it can be noted from the XRD analysis and the crystallite size that along with the calcination temperature, the time period and the heating rate required for calcination is also important to yield pure and smaller nanoparticles.

Crystallinity of selected MgO sample.
4.2 Functional Group Analysis
The functional groups present in the selected, calcinated MgO sample was characterized using FTIR spectroscopy and the result are listed in Table 6 and Fig. 8. The FTIR spectra shows peaks at 3695.1, 3463.6, 1435.8, 1350.4 and 1047.6 cm–1 indicating O–H stretch, C–O–H bending and C–O stretch vibrational mode for the presence of alcohols and carboxylic acid. The peaks at 2921, 1737.9, 1696.4 and 1642.2 cm–1 which are due to C–H stretch, C=O stretch and –C=C- vibrations support the presence of alkanes, α, β – unsaturated esters and aldehydes, saturated aliphatics, ketones and alkenes in the sample respectively. The peaks at 867.6, 834.1 and 687.4 cm–1 are due to MgO vibrations [37, 63–65]. The peaks at 2430.3, 2320.3, 1841.5 and 1540.7 cm–1 are not designated to any functional groups and thus are categorised as impure peaks. The presence of these impure peaks may be due to the polycrystalline nature of the sample as mentioned in XRD analysis. Literatures suggested that increase in calcination time will reduce these impurity peaks [66]. The peak at 867.6 and 834.1 cm–1 are due to distinct MgO vibrations attributed to the periclase phase of MgO [37]. The presence of alcohol functional group traces represents the existence of magnesium hydroxide in the polycrystalline sample. The presence of these intermediate hydroxide and peroxide compounds leads to the vibrational modes with C–O stretch and –C=C– stretch. Hence, it can be noted that carboxylic and alcohol group may act as capping agent in the formation of MgO nanoparticles which controls their grain size [67]. The additional oxygen and hydrogen molecules which are present due to inadequate calcination temperature, may settle in the interstital position of these capping agents. As the calcination time is short for complete reaction, the additional oxygen and hydrogen molecules bind with the functional groups of the capping agent which leads to the formation of a polycrystalline MgO sample.

FTIR spectra of selected MgO sample.
4.3 Morphology Studies
Figure 9 shows the morphology of particles formed in sample B calcinated at 500°C. In Fig. 9a, it can be noted that the particles are agglomerated and mostly the particles are spherical in shape with certain irregular shaped particles. The size of the particles were observed to be between 30–45 nm which in compliance with the DLS and XRD crystallite size data. Figures 9b, 9c shows the morphology of a single particle which is 30 nm sized spherical particle. Eventhough, the crystallinity of the sample B calcinated at 500°C is observed to contain impure magnesium oxide crytals, the size and shape of particles are definite. The impurity and the agglomeration can be reduced by altering the time and heating rate of calcination process [68–70]. Mastuli et al. (2014) prepared MgO nanoparticles using magnesium acetate as precursor and tartaric acid as gelling agent. The sol–gel product was calcinated at 950°C for 36 h to yield different shapes of MgO nanoparticles [71]. In another work by Tamilselvi et al. (2013), 30–50 nm sized MgO nanoparticles prepared by using magnesium nitrate as precursor, sodium hydroxide as reducing agent and calcinated at 500°C for 4 h [63]. Thus, it is evident that alteration in the time and heating rate of calcination process will yield pure and uniform morphology of MgO nanoparticles.

Electron micrograph of sample B calcinated at 500°C (a) Nanoparticles at 100 nm scale, (b) Single nanoparticle at 100 nm scale and (c) Zoomed image of single nanoparticle at 50 nm scale.
5 CONCLUSIONS
In the present work, MgO nanoparticles were synthesized by sol–gel method using magnesium nitrate as the precursor, different combinations of gelling agents, and ethanol as the solvent. X-ray diffraction method and thermal analysis were used to characterize the crystallinity of sol–gel products and to select optimal calcination temperatures for MgO nanoparticles formation. The average particle sizes of the calcinated samples were determined by dynamic light scattering. It was demonstrated that crystallinity plays a major role in determining the thermal degradation characteristics, calcination temperature, and the particulate size of MgO nanoparticles produced by sol–gel synthesis. Also, different types and compositions of gelling agents were demonstrated to affect the crystallinity of the sol–gel product. Sample B which was calcinated at 500°C was selected for further analysis as the average particle size was found to be below 100 nm compared to other samples. Thus, the sample was subjected to XRD, FTIR and TEM analysis which reveals that the calcination time and heating rate has to be prolonged to obtain pure, uniform morphology of MgO nanoparticles. The current work reveals the effect of gelling agent in the formation of sol–gel product which eventually affects the calcination temperature required to form nanoparticles. Further, it also discloses the effect of calcination profiles in modifying the shape, size and agglomeration which will help to synthesize uniform sized nanoparticles with less agglomeration.
REFERENCES
Kajihara, K., J. Asian Ceram. Soc., 2013, vol. 1, p. 121.
Rideal, E. and Davies, J., Interfacial Phenomena, London: Academic Press, 1963.
Hench, L.L. and West, J.K., Chem. Rev., 1990, vol. 90, p. 33.
Niederberger, M., Acc. Chem. Res., 2007, vol. 40, p. 793.
Mackenzie, J.D., J. Non-Cryst. Solids, 1988, vol. 100, p. 162.
Rao, C. and Raveau, B., Annu. Rev. Phys. Chem., 1989, vol. 40, p. 291.
Niederberger, M. and Pinna, N., Metal Oxide Nanoparticles in Organic Solvents: Synthesis, Formation, Assembly and Application, Heidelberg: Springer, 2009.
Stoimenov, P.K., Klinger, R.L., Marchin, G.L., and Klabunde, K.J., Langmuir, 2002, vol. 18, p. 6679.
Veldurthi, S., Shin, C.-H., Joo, O.-S., and Jung, K.-D., Microporous Mesoporous Mater., 2012, vol. 152, p. 31.
Krishnamoorthy, K., Moon, J.Y., Hyun, H.B., Cho, S.K., and Kim, S.-J., J. Mater. Chem., 2012, vol. 22, p. 24610.
Gulková, D., Šolcová, O., and Zdražil, M., Microporous Mesoporous Mater., 2004, vol. 76, p. 137.
Climent, M.J., Corma, A., Iborra, S., and Mifsud, M., J. Catal., 2007, vol. 247, p. 223.
Faghihi-Sani, M.-A. and Yamaguchi, A., Ceram. Int., 2002, vol. 28, p. 835.
Ouraipryvan, P., Sreethawong, T., and Chavadej, S., Mater. Lett., 2009, vol. 63, p. 1862.
Makhluf, S., Dror, R., Nitzan, Y., Abramovich, Y., Jelinek, R., and Gedanken, A., Adv. Funct. Mater., 2005, vol. 15, p. 1708.
Bertinetti, L., Drouet, C., Combes, C., Rey, C., Tampieri, A., Coluccia, S., and Martra, G., Langmuir, 2009, vol. 25, p. 5647.
Martinez-Boubeta, C., Balcells, L., Cristòfol, R., Sanfeliu, C., Rodríguez, E., Weissleder, R., Lope-Piedrafita, S., Simeonidis, K., Angelakeris, M., and Sandiumenge, F., Nanomed.: Nanotechnol., Biol., Med., 2010, vol. 6, p. 362.
Chalkidou, A., Simeonidis, K., Angelakeris, M., Samaras, T., Martinez-Boubeta, C., Balcells, L., Papazisis, K., Dendrinou-Samara, C., and Kalogirou, O., J. Magn. Magn. Mater., 2011, vol. 323, p. 775.
Di, D.-R., He, Z.-Z., Sun, Z.-Q., and Liu, J., Nanomed.: Nanotechnol., Biol., Med., 2012, vol. 8, p. 1233.
Mastuli, M.S., Kamarulzaman, N., Nawawi, M.A., Mahat, A.M., Rusdi, R., and Kamarudin, N., Nanoscale Res. Lett., 2014, vol. 9, p. 1.
Rao, J.P. and Geckeler, K.E., Prog. Polym. Sci., 2011, vol. 36, p. 887.
Jeevanandam, J., San Chan, Y., and Danquah, M.K., Biochimie, 2016, vol. 128, p. 99.
Ding, Y., Zhang, G., Wu, H., Hai, B., Wang, L., and Qian, Y., Chem. Mater., 2001, vol. 13, p. 435.
Esmaeili, E., Khodadadi, A., and Mortazavi, Y., J. Eur. Ceram. Soc., 2009, vol. 29, p. 1061.
Nemade, K. and Waghuley, S., Int. J. Met., 2014, vol. 2014, p. 389416.
Mastuli, M.S., Kamarulzaman, N., Nawawi, M.A., Mahat, A.M., Rusdi, R., and Kamarudin, N., Nanoscale Res. Lett., 2014, vol. 9, p. 1.
Suresh, S., J. Ovonic Res., 2014, vol. 10, p. 205.
Demirci, S., Öztürk, B., Yildirim, S., Bakal, F., Erol, M., Sancakoğlu, O., Yigit, R., Celik, E., and Batar, T., Mater. Sci. Semicond. Process., 2015, vol. 34, p. 154.
Mastuli, M.S., Ansari, N.S., Nawawi, M.A., and Mahat, A.M., APCBEE Proc., 2012, vol. 3, p. 93.
Vatsha, B., Tetyana, P., Shumbula, P.M., Ngila, J.C., Sikhwivhilu, L.M., and Moutloali, R.M., J. Biomater. Nanobiotechnol., 2013, vol. 4, p. 365.
Uche, D.O.V., Adv. Appl. Sci. Res., 2013, vol. 4, p. 506.
Zhang, J.-J., Ning, J.-W., Liu, X.-J., Pan, Y.-B., and Huang, L.-P., Mater. Res. Bull., 2003, vol. 38, p. 1249.
Kareiva, A., Karppinen, M., and Niinisto, L., J. Mater. Chem., 1994, vol. 4, p. 1267.
Thirunakaran, R., Kim, K.-T., Kang, Y.-M., and Young-Lee, J., Mater. Res. Bull., 2005, vol. 40, p. 177.
Park, S.H. and Sun, Y.-K., J. Power Sources, 2003, vols. 119–121, p. 161.
Danks, A., Hall, S., and Schnepp, Z., Mater. Horiz., 2016, vol. 3, p. 91.
Kumar, A. and Kumar, J., J. Phys. Chem. Solids, 2008, vol. 69, p. 2764.
Jaison, J., Balakumar, S., and Chan, Y., IOP Conf. Ser.: Mater. Sci. Eng., 2015, vol. 78, p. 012005.
Jeevanandam, J., Chan, Y.S., and Danquah, M.K., ChemistrySelect, 2017, vol. 2, p. 10393
Gupta, P.K., J. Non-Cryst. Solids, 1996, vol. 195, p. 158.
Lu, K., Mater. Sci. Eng., R, 1996, vol. 16, p. 161.
Braibanti, A., Tiripicchio, A., Camellini, M., Lanfredi, A., and Bigoli, F., Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, vol. 25, p. 354.
Simonsen, O., Mogelmosea, M.-L.H., and Courb, A.I., Acta Chem. Scand., 1999, vol. 53, p. 432.
Szabó, T., Tombácz, E., Illés, E., and Dékány, I., Carbon, 2006, vol. 44, p. 537.
Zallen, R., The Physics of Amorphous Solids, John Wiley and Sons, 1983.
Hilden, L.R. and Morris, K.R., J. Pharm. Sci., 2004, vol. 93, p. 3.
Livage, J. and Sanchez, C., J. Non-Cryst. Solids, 1992, vol. 145, p. 11.
Doreswamy, B., Mahendra, M., Sridhar, M., Prasad, J.S., Varughese, P., George, J., and Varghese, G., Mater. Lett., 2005, vol. 59, p. 1206.
Sato, N., Quitain, A.T., Kang, K., Daimon, H., and Fujie, K., Ind. Eng. Chem. Res., 2004, vol. 43, p. 3217.
Keely, W. and Maynor, H.W., J. Chem. Eng. Data, 1963, vol. 8, p. 297.
Tanaka, K., Capule, M.F., and Hisanaga, T., Chem. Phys. Lett., 1991, vol. 187, p. 73.
Yu, J.-G., Yu, H.-G., Cheng, B., Zhao, X.-J., Yu, J.C., and Ho, W.-K., J. Phys. Chem. B, 2003, vol. 107, p. 13871.
Zhang, W.-X., Wang, Y.-Z., and Sun, C.-F., J. Polym. Res., 2007, vol. 14, p. 467.
Golberg, D., Bando, Y., Fushimi, K., Mitome, M., Bourgeois, L., and Tang, C.-C., J. Phys. Chem. B, 2003, vol. 107, p. 8726.
Christodoulou, C., Schlup, J., and Hadjipanayis, G., J. Appl. Phys., 1987, vol. 61, p. 3760.
Pang, Y.X. and Bao, X., J. Eur. Ceram. Soc., 2003, vol. 23, p. 1697.
Yu, J. and Wang, B., Appl. Catal., B, 2010, vol. 94, p. 295.
Gibson, I., Rehman, I., Best, S., and Bonfield, W., J. Mater. Sci.: Mater. Med., 2000, vol. 11, p. 799.
Pokroy, B., Fitch, A.N., Lee, P.L., Quintana, J.P., El’ad, N.C., and Zolotoyabko, E., J. Struct. Biol., 2006, vol. 153, p. 145.
Keshmiri, M., Mohseni, M., and Troczynski, T., Appl. Catal., B, 2004, vol. 53, p. 209.
Wang, L. and Muhammed, M., J. Mater. Chem., 1999, vol. 9, p. 2871.
Demir, M.M., Gulgun, M.A., Menceloglu, Y.Z., Erman, B., Abramchuk, S.S., Makhaeva, E.E., Khokhlov, A.R., Matveeva, V.G., and Sulman, M.G., Macromolecules, 2004, vol. 37, p. 1787.
Tamilselvi, P., Yelilarasi, A., Hema, M., and Anbarasan, R., Nano Bull., 2013, vol. 2, p. 130106.
Rezaei, M., Khajenoori, M., and Nematollahi, B., Powder Technol., 2011, vol. 205, p. 112.
Song, G., Ma, S., Tang, G., and Wang, X., Colloids Surf., A, 2010, vol. 364, p. 99.
Santander, N., Das, S.R., Majumder, S.B., and Katiyar, R.S., Surf. Coat. Technol., 2004, vol. 177, p. 60.
Vignesh Subramanian, I.B., Pranav, R. and Rajendran, N., J. Chem. Pharm. Res., 2015, vol. 7, p. 116.
Rhodes, W.H., J. Am. Ceram. Soc., 1981, vol. 64, p. 19.
Yeh, M.Y., Lin, C., Vu, C.T., Hsu, K.F., Lee, S.Y., Li, W.R., and Yen, C.-F., Mater. Res. Bull., 2017, vol. 90, p. 182.
Namvar, N., Salahinejad, E., Saberi, A., Baghjeghaz, M.J., Tayebi, L., and Vashaee, D., Ceram. Int., 2017, vol. 43, no. 16, pp. 13781–13785.
Mastuli, M.S., Kamarulzaman, N., Nawawi, M.A., Mahat, A.M., Rusdi, R., and Kamarudin, N., Nanoscale Res. Lett., 2014, vol. 9, p. 134.
FUNDING
The authors wish to acknowledge Curtin University, Malaysia for financial support through the Curtin Sarawak Postgraduate Research Scholarship (CSPRS) scheme.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jaison Jeevanandam, San Chan, Y. & Danquah, M.K. Effect of Gelling Agent and Calcination Temperature in Sol–Gel Synthesized MgO Nanoparticles. Prot Met Phys Chem Surf 55, 288–301 (2019). https://doi.org/10.1134/S2070205119020114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S2070205119020114