
Abstract
The feasibility of obtaining ruthenium oxide nanoparticles on a carbon support by pyrolysis of polymeric ruthenium complexes is shown. The performance efficiency of such a catalyst considerably surpasses the efficiency of catalysts synthesized using monomer ruthenium compounds and is comparable with figure of merit values reported for the best samples of hydrogen evolution reaction catalysts in alkaline media, while the specific consumption of the precious metal in this catalyst is much lower.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Due to growing concerns regarding an approaching energy crisis and environmental pollution, huge efforts are being put into the development of green energy sources. As a clean, sustainable, and energy-dense (120 MJ/kg) compound [1], molecular hydrogen is considered to be a next-generation energy source and the basis of hydrogen power engineering [1, 2].
At present, hydrogen is mostly produced by steam reforming of natural gas. That being so, a fossil fuel is used and carbon dioxide, a culprit of greenhouse effect [3], is released. Therefore, as a carbon-free process of H2 production, electrochemical water splitting, attracts considerable interest [4, 5]. Developing efficient electrocatalysts for water splitting is thought to be one most serious problem in implementation of hydrogen production on an industrial scale [1, 6].
Platinum is known to be the most efficient catalyst for electrochemical hydrogen evolution reaction (HER), especially in acidic solutions. In the meantime, the efficiency of a platinum catalyst in neutral and alkaline solutions drops by 2–3 orders of magnitude because of slowness of the water dissociation step [7], with a platinum load of 300–500 μg/cm2 [2].
Ruthenium, which is fivefold cheaper than platinum [8], attracts attention of researches as a HER catalyst in alkaline media: the energy of Ru–H bond (272 kJ/mol) is nearly the same as that for platinum, and the activation energy of water dissociation on ruthenium is half as high as that on platinum [2]. The factors indicated above considerably promote the Volmer step of HER in alkaline media [9].
Thermolysis of organic ruthenium precursors is one key method for preparing ruthenium-containing catalysts [2]. Meanwhile, synthesis methods described in the absolute majority of literature sources begin with preparation of an organic matrix, and then Ru3+ ions are incorporated into it (typically, a solution of ruthenium(III) chloride is used for this), followed by pyrolysis of the resulting material [10–14].
Earlier, we patented [15] and then published [16] a method for nanocatalyst synthesis, in which films of polymer metal complexes are used as precursors to catalytically active particles. Such films are deposited onto an electrode by electropolymerization of corresponding monomeric compounds and subjected to pyrolysis. Catalyst syntheses using polymer−metal films have a number of advantages over other methods used:
–electrochemical polymerization and subsequent thermal treatment of a polymer enable us to obtain nanocatalysts containing a transition metal directly on the electrode surface;
–due to strong adhesion of the polymer film to the electrode surface, the catalyst resulting from this film has good adhesion to the surface, which spares us the use of a binder;
–the proposed method ensures uniform coverage of the electrode surface with the catalyst, while the surface concentration of catalytically active sites, as well as their size, can be adjusted by varying the thickness of polymeric film, i.e., by polymerization conditions;
–by copolymerization of complexes with different metal sites, we can create complex catalytic systems, e.g., bifunctional catalysts.
The aim of this work is to apply a method of pyrolysis of ruthenium-containing polymer complexes to synthesize nanoscale catalysts and investigate their HER activity in alkaline solutions.
EXPERIMENTAL
Regardless of catalyst synthesis technique used, nonactivated carbon cloth Kynol CC-509 (specific surface area, 0.03 m2/g) was used an electrode support material, the electrode area being 1.266 cm2.
The catalyst-modified electrodes for HER in alkaline media were prepared using three catalyst precursors:
–a polymer complex of Ru(II) and 1,10-phenanthroline (poly-[Ru(Phen)3]2+) (1) (the key electrodes used in our study);
–the monomer complex Ru(5–Cl–Phen)3]2+ (2) (for electrodes used for obtaining comparative parameters of catalysts based on polymeric and monomer complexes);
–ruthenium(III) chloride (3) (for electrodes used for obtaining comparative parameters of catalysts based on the considered polymers and a commonly used precursor [10–14]).
For synthesizing polymer 1, the initial complex [Ru(5–Cl–Phen)3](BF4)2 was prepared using data [17]. The polymerization on working electrode was carried out, using a three-electrode cell, in a 2 mM solution of the initial complex in acetonitrile (AN) that also contained 0.1 mol/L of Et4NBF4 (Aldrich). The reference electrode was a nonaqueous Ag+/Ag (MW-1085, BASi) electrode filled with a 5 mM AgNO3 solution, and a piece of carbon cloth (~5 cm2) was used as the auxiliary electrode. Polymerization parameters are described at greater length in the next section.
After the polymerization was complete, the electrodes were carefully washed in AN and dried under argon. Thermal decomposition of polymers was carried out in a PTK-1.2-40 tubular furnace (Teplopribor, Russia) at 500°C in argon or at 350°C in air for 1 h (the temperature was maintained with a precision of ±1°C).
When using precursors 2 and 3, we applied them on the working electrode in a dissolved form: 200 μL of a 8.7 mM solution of [Ru(5–Cl–Phen)3](BF4)2 in AN or 50 μL of a 0.1 M aqueous RuCl3 solution (Aldrich), respectively.
After the precursor was applied, the electrodes were dried at 120°C for 1 h. The precursors were subjected to pyrolysis similarly to case 1.
The mass of catalytic formations was determined by weighing the initial working electrode and the working electrode after being subjected to pyrolysis; the accuracy of weighing was 0.1 mg. From control experiments, we found out that the mass of carbon cloth itself changed by no more than 1% as a result of thermal treatment in argon.
The catalysts prepared were investigated by scanning electron microscopy (SEM) using a Zeiss Merlin microscope equipped with an INCA X-act attachment (Oxford Instruments) for X-ray microanalysis.
HER studies with the catalyst-modified electrodes were carried out at room temperature in a cell with separate compartments for the working, auxiliary, and reference electrodes. A deaerated aqueous 0.2 M KOH solution was used as the electrolyte. An Ag/AgCl reference electrode filled with an aqueous 3 M NaCl solution was brought in contact with the working electrode space via a Luggin–Haber capillary. All potentials in aqueous solutions are henceforth quoted with respect to the indicated reference electrode; in particular, the potential of reversible hydrogen electrode in this solution is –0.994 V. A 2 cm2 piece of EQ-bcnf-16m-2 porous nickel foam (MTI) was used as the auxiliary electrode. Polarization of the working electrode was performed potentiodynamically in the range of –0.9 to –1.1 V at a rate of 5 mV/s using a VSP potentiostat (BioLogic Science Instruments).
RESULTS AND DISCUSSION
It is known that polymer films of poly-[Ru(Phen)3]2+ can be obtained on the surface of a glassy carbon electrode in a 2 mM solution of [Ru(5–Cl–Phen)3](BF4)2 in AN either by scanning the potential cyclically from –1 to –2 V vs. Ag+/Ag reference electrode [17] or under potentiostatic conditions by poising the electrode (in the same solution) at –1.8 ± 0.1 V [18]. To deposit a polymer onto the carbon cloth, we used pulsed potentiostatic deposition [19] at –1.8 V, the pulse duration and the pulse-off time being 1 and 10 s, respectively. During a short period of electrode polarization, the polymer forms on the surface of filaments in the carbon cloth, which results in a decrease in the monomer concentration near the electrode and, therefore, a lowering of the rate of polymerization process, especially on internal filaments of the carbon material. A long pause after each polarization pulse enables partial or complete recovery of the initial monomer concentration profile inside the cloth, which ensures effective formation of the polymer on the internal cloth filaments during the next pulse of electrode polarization. A SEM image of a polymer deposited on the surface of working electrode, after the electrode was washed and then dried in vacuum, is shown in Fig. 1a. By varying the number of polymerization pulses, we were able to change the mass of polymer deposited on the electrode surface: during 50 pulses, 0.9 mg was deposited; during 100 pulses, 1.1 mg; and during 150 pulses, 1.3 mg (or, in terms of Ru load, we had 110, 135, and 160 μg of Ru). Increasing the number of pulses further did not result in a considerable increase in the mass of deposited polymer, which is likely due to growing resistance through the film [18].

SEM images of a poly-[Ru(Phen)3]2+ film formed on the surface of working electrode (150 polymerization pulses): (a) as deposited and (b) after pyrolysis at 500° in argon.
Thermal treatment under inert atmosphere did not lead to complete decomposition of the polymer even at 500°C: the film thickness decreased and the number and depth of cracks grew, but the organic ligand environment around the ruthenium was preserved, as can be appreciated from Fig. 1b.
However, the organic component can be completely removed by pyrolysis in air at temperatures as low as 350° (Fig. 2). The X-ray microanalysis of pyrolysis products showed the presence of ruthenium and oxygen, and, by taking into account the shape of crystals [20], this can be interpreted as the presence of a stable form of ruthenium(IV) oxide on the surface.

SEM images of pyrolysis products of a poly-[Ru(Phen)3]2+ film (150 polymerization cycles) on the surface of working electrode after pyrolysis at 350°C in air: (a) particles of a ruthenium-containing catalyst (average size of 70 ± 10 nm) and (b) distribution of particles on the surface of an individual filament in carbon cloth.
For comparative testing of efficiency in HER, we selected the following types of electrodes:
–electrode with a catalyst based on the polymer ruthenium complex synthesized by running 150 polymerization cycles, which was subjected to pyrolysis at 350° in air and contains 120 μg of Ru per 1 cm2 of the surface area. This relatively high catalyst load was chosen because the best catalytically active electrodes developed so far contain 200–400 μg of Ru per cm2 [2];
–electrode with a catalyst based on the monomer ruthenium complex, which was subjected to pyrolysis at 350°C in air and contains 120 μg of Ru per 1 cm2 of the geometric surface area)
–electrode with a catalyst based on ruthenium(III) chloride, which was subjected to pyrolysis at 350° in air and contains 400 μg of Ru per 1 cm2 of the geometric surface area (similar to data [2]).
Before registering voltammograms, all the electrodes were subjected to an activation process, which consisted in 30-min long polarization at a current of –30 mA. Activation of the oxide layer is known to be a key element to the electrocatalytic behavior of RuO2 in HER: it boosts the catalyst performance severalfold [20]. Typically, activation is thought to be related to incomplete reduction of ruthenium oxide to form an oxyhydroxy complex [20].
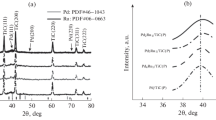
HER voltammograms for the three types of electrodes described above are shown in Fig. 3. The catalyst performance efficiency was expressed in the units of cathodic current per 1 mg of ruthenium at a potential of –1.1 V. All the voltammograms were fully reproducible on the time scale of the experiment (i.e., a few hours).

(Color online) Voltammograms for the HER registered at a potential sweep rate of 5 mV/s in an aqueous 0.2 M KOH solution on an electrode (area of 1.266 cm2) modified with the ruthenium-containing catalyst obtained by pyrolysis of (1) polymer, (2) monomer complex, and (3) RuCl3. The current is normalized to the ruthenium mass in the catalyst.
The best catalytic activity in HER was registered for the electrodes for which the catalyst was obtained by pyrolysis of the polymer. Preliminary activation of such electrodes gives the most substantial result: the current at a potential of –1.1 V increases by more than 2.5-fold. The performance efficiency of catalysts prepared by pyrolysis of a monomer complex, all other conditions being equal, was 30% lower, and for the catalysts based on ruthenium(III) chloride it was around fourfold lower. It is likely that the polymer matrix hinders agglomeration of ruthenium oxide nanoparticles during pyrolysis, whereas SEM images of the electrodes prepared using monomer precursors reveal the presence of nanoparticle agglomerates with a size up to a few microns.
The following key figures of merit used in literature were determined from voltammetry measurements for the catalyst prepared by pyrolysis of the polymer complex: hydrogen overvoltage η at a current density of 10 mA/cm2 is 55 mV, Tafel slope dη/d(lgi) is 60 mV, and turnover frequency (TOF; the number of H2 moles per mole of catalyst per second) is 0.1 s–1. We note that the results reported here are for a diluted (0.2 M) alkali solution, whereas as the OH– concentration increases the HER overvoltage and Tafel slope decrease, while TOF grows. Of course, for practical application of the catalysts developed here, long-term performance testing in concentrated alkali solutions at high temperatures is required, and such experiments are currently under way in the Ioffe institute.
CONCLUSIONS
We showed the feasibility of obtaining ruthenium oxide nanoparticles on a carbon support by subjecting a polymer complex of ruthenium to pyrolysis. The performance efficiency of such a catalyst surpasses considerably the efficiency of catalysts synthesized using monomer ruthenium compounds and is comparable with figures of merit reported for the best HER catalysts in alkaline media [2], while the specific consumption of the precious metal for this catalyst is much lower.
REFERENCES
Electrochemical Water Electrolysis: Fundamentals and Technologies (CRC, Boca Raton, FL, 2020).
S. Y. Bae, J. Mahmood, I. Y. Jeon, and J. B. Baek, Nanoscale Horiz. 5, 43 (2020). https://doi.org/10.1039/c9nh00485h
M. Zeng and Y. Li, J. Mater. Chem. A 3, 14942 (2015). https://doi.org/10.1039/C5TA02974K
Y. Liu, G. Yu, G.-D. Li, et al., Angew. Chem. 54, 10752 (2015). https://doi.org/10.1002/anie.201504376
J. Mahmood, M. A. R. Anjum, S.-H. Shin, et al., Adv. Mater. 30, 1805606 (2018). https://doi.org/10.1002/adma.201805606
Y. Zheng, Y. Jiao, M. Jaroniec, et al., Angew. Chem. 54, 52 (2015). https://doi.org/10.1002/anie.201407031
D. Strmcnik, P. P. Lopes, B. Genorio, et al., Nano Energy 29, 29 (2016). https://doi.org/10.1016/j.nanoen.2016.04.017
Y. Zheng, Y. Jiao, Y. Zhu, et al., J. Am. Chem. Soc. 138, 16174 (2016). https://doi.org/10.1021/jacs.6b11291
Handbook of Functionalized Nanomaterials for Industrial Applications (Elsevier, Amsterdam, 2020).
J. Mahmood, F. Li, S.-M. Jung, et al., Nat. Nanotechnol. 12, 441 (2017). https://doi.org/10.1038/NNANO.2016.304
B. Lu, L. Guo, F. Wu, et al., Nat. Commun. 10, 631 (2019). https://doi.org/10.1038/s41467-019-08419-3
J. Zhang, P. Liu, G. Wang, et al., J. Mater. Chem. A 5, 25314 (2017). https://doi.org/10.1039/C7TA08764K
Y. Li, L. A. Zhang, Y. Qin, et al., ACS Catal. 8, 5714 (2018). https://doi.org/10.1021/acscatal.8b01609
Q. Song, X. Qiao, L. Liu, et al., Chem. Commun. 55, 965 (2019). https://doi.org/10.1039/c8cc09624d
RF Patent No. 2675582 C2, Byull. Izobret., No. 35 (2018).
M. V. Novozhilova, Yu. S. Danilova, M. P. Karushev, A. M. Timonov, V. V. Malev and O. V. Levin, Russ. J. Electrochem. 54, 769 (2018). https://doi.org/10.1134/S102319351810004X
O. Fussa-Rydel, H. T. Zhang, J. T. Hupp, et al., Inorg. Chem. 28, 1533 (1989). https://doi.org/10.1021/ic00307a022
A. N. Borisov, A. M. Timonov, V. A. Timofeev, and G. A. Shagisultanova, Russ. J. Inorg. Chem. 41, 435 (1996).
I. A. Chepurnaya, S. A. Logvinov, M. P. Karushev, A. M. Timonov, and V. V. Malev, Russ. J. Electrochem. 48, 538 (2012). https://doi.org/10.1134/S1023193512040040
H. Over, Chem. Rev. 112, 3356 (2012). https://doi.org/10.1021/cr200247n
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by A. Kukharuk
Rights and permissions
About this article

Cite this article
Polozhentseva, Y.A., Bykov, V.A., Timonov, A.M. et al. Nanostructured Ruthenium Oxide for Electrocatalysis of Hydrogen Evolution Reaction in Aqueous Alkaline Solutions. Nanotechnol Russia 15, 730–734 (2020). https://doi.org/10.1134/S1995078020060130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1995078020060130