
Abstract
The paper presents the results of calculations of the dynamics of ozone destruction in the lower stratosphere, carried out taking into account the heterogeneous chemical reactions (HCRs) proceeding at a rate of w(–O3)HCRs, cm–3 s–1, with the participation of particles of the Junge layer (background aerosol). The dramatic decline in w(–O3)HCRs found in the calculations at altitudes less than 16 km in comparison with the rate of ozone loss calculated with the participation of gaseous chemical reactions (w(–O3)) indicates the inhibitory role of aerosol particles. This is due to the capture of N2O5 molecules from the air by aerosol particles. Their rapid runoff entails a sharp decrease in the concentrations of components of the NOx family in air, as well as a less pronounced decrease in the concentrations of components of the HOx, and Ox families involved in the destruction of ozone. At higher altitudes of 16 to 22 km, magnitude w(–O3)HCRs, in contrast, turns out to be slightly higher than w(–O3); and it affects the acceleration of the destruction process with the components of the HOx and ClOx families. The increased level of their concentrations and rates of reactions with ozone is due to the reduced content of the components of the NOx family in the air. This positive effect of the HCRs involving aerosol particles in w(–O3)HCRs practically degenerates, but at even higher altitudes. This is due to the decrease in the content of aerosol particles and the acceleration of the photodissociation of molecules N2O5(g) \(\xrightarrow{{h{{\nu }}}}\) NO3(g) + NO2(g), which on the whole is accompanied by the suppression of the process of their capture by particles.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Earlier [1], when considering the influence of particles of the Junge layer [2] on the lifetime of “odd oxygen” in the lower stratosphere, we noted the important role of heterogeneous chemical reactions (HCRs) [3–5] in a complex cycle of atmospheric chemical processes involving components of the Ox family. The influence of the particles of the Junge layer on the rate of ozone destruction is indirectly indicated by the data of field observations of the concentrations of the components of the families participating in the destruction of ozone in the middle latitudes in the Stratospheric Photochemistry Aerosols and Dynamics Expedition (SPADE) field experiments [6]. In this regard, in [7], we pointed out that it is HCRs with the participation of particles of this layer that are the reason for the negative trend in the ozone concentration in the lower stratosphere during the so-called ozone crisis (the end of the 20th century). The aim of this study is to carry out numerical calculations taking into account the effect of sulfate aerosol on the rate of ozone destruction at altitudes corresponding to the arrangement of particles of the Junge layer in the lower stratosphere: 10–25 km.
ChemWG MODEL
In this work, as in [7], the effect of HCRs with the participation of particles of the Junge layer on the concentration of small components in the lower stratosphere and the rates of reactions with their participation was judged using the previously constructed 0‑dimensional model (ChemWG). In [8], the local ozone destruction in the lower stratosphere at high latitudes was considered in a similar way (68 ± 10)° north latitude with the participation of large (at least 5 μm) particles of nitric acid trihydrate (NAT rocks). The ChemWG model, which includes a block of chemical reactions in the gas phase, similar to the SOCRATES model [9], as well as blocks of mass transfer processes at the gas–drop interface and liquid-phase reactions in drops, is described in [7]. The numerical calculations were performed for a unit volume of gas with droplets uniformly distributed over its volume (r = 0.1 μm) at altitudes ranging from 10 to 25 km. The counting time was ≤3 × 106 s (≈30 days). At the same time, as in the SOCRATES model, the content of trace impurities was calculated taking into account their daily changes. The calculations took into account the diffusion limitations of the liquid-phase reactions in the volume of the aerosol particles.
Based on the results of the consideration, it was found that the main role in these conditions is played by the N2O5 hydrolysis processes and to a much lesser extent ClONO2 hydrolysis processes:
Both of these processes proceed rapidly in the droplets' subsurface layer, and their speeds are proportional to their specific surface area: 3L/r. Here L is the volumetric content of the droplet moisture in the gas (cm3 aerosol/cm3 air).
The specific values of the volume/surface of the sulfate aerosol particles in the lower stratosphere necessary for calculating the rates of these HCRs were borrowed from the data of field experiments that measured the sulfuric acid content in the lower stratosphere [10]. The physicochemical properties of supercooled drops (the sulfuric acid content is 50–78% wt %, density is 1.4–1.8 g/ mL, activity of water and free protons are, respectively, ∼2 × 10–3–0.3 and 3 × 102–3 × 105 mol/L, and others) were calculated using the Atmospheric Inorganic Model (AIM) [11]. The calculations also used data on the solubility of reservoir chlorine-containing gases (HCl, ClNO3) and N2O5, which were obtained in experiments (the wetted wall flow tube technique [12] and others) to study the dynamics of gas molecules being captured by supercooled solutions of sulfuric acid, as well as data on their viscosity and kinetics of low-temperature liquid-phase chemical reactions [13]. The altitude profiles of the concentrations of individual components in the gas phase (xi, cm–3), as well as temperature and relative humidity (RH) at the given geographic point were calculated using a two-dimensional interactive SOCRATES model. The calculation results presented below correspond to the middle latitudes (50° N) and summer time (June 1995).
CALCULATION RESULTS AND DISCUSSION
According to the results of the calculations, it was found that, despite the low solubility of most components of the families under consideration, their altitude profiles exhibit significant changes when taking into account the capture of gas molecules N2O5 and СlONO2 by aerosol. Thus, the concentration of NO2, one of the components of the NOx-family, at altitude ≤17 km turns out to be much lower (by an order of magnitude [1]) in comparison with those calculated by us in the absence of HCRs. The changes in the concentrations of N2O5 , the predecessor of NO2, turn out to be even more noticeable: N2O5(g) \(\xrightarrow{{h{{\nu }}}}\) NO3(g) + NO2(g). Their values turn out to be three to four orders of magnitude lower than the calculated values [N2O5] neglecting hydrolysis (1). The decrease by factors of 3 to 4 at these altitudes in the concentrations of OH and HO2 radicals—components of the NOx-family—is also related to the quick capture of sulfate aerosol by N2O5 particles near the lower boundary of the Junge layer. Their decline is caused by the decrease in the concentration of HNO3 due to both the dissolution and decrease in [OH] and [NO2] and, as a result, suppression of the photodissociation of nitric acid vapors. The decline at these altitude of the concentration of ClOx, a component of the ClO family, participating in the destruction of ozone, is also related to the capture of chlorinitrate by aerosol particles according to reaction (2).
As noted in [1], the calculated altitude profiles of the concentrations of the components of the NOx, HOx, and ClOx families repeat the altitude profile of the distribution of the mass concentration of particles of the Junge layer in the lower stratosphere. In this case, however, the maximums of the OH and ClO concentrations are at somewhat higher altitudes (by 1–3 km) than the minimum of the NO2 concentration. In the case of the OH radical, the upward shift in the maximum of their concentration is due to the increase in the ozone content; and in the case of ClO, the content of reservoir chlorine-containing gases (HCl, ClONO2), which are precursors of ClO, increases with altitude above the underlying surface, since chlorofluorocarbons, which are located at higher altitudes, are the main source of HCl and ClONO2.
We find evidence of changes in the concentrations of the components of the families participating in the destruction of ozone caused by aerosol particles by considering the data of the SPADE field experiments cited above [6]. Thus, at a altitude of ∼19 km (∼50° north latitude, ≈67 mbar, T ≈ 216 K, mid-May 1993), according to these data, the NO2 content fell by almost one-third, the concentration of OH and HO2 increased by approximately 30–50%, and the ClO concentration approximately quadrupled. In this case, the concentrations of these components, calculated in the approximation of the absence of HCRs with particles, were considered as the baseline level of their content (i.e., in the absence of particles from the Junge layer). A similar increase is demonstrated by the concentrations of OH and ClO radicals according to the data of our calculations [1].
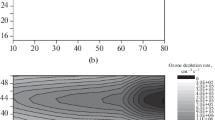
Figure 1 shows the vertical profiles of the calculated partial rates of ozone destruction in the catalytic cycles of HOx, NOx, ClOx, and Ox, as well as the vertical profile of their total velocity. When calculating them in each of these cycles, those of them that are characterized by the lowest speed and limit the dynamics of the cycle as a whole were considered as links in the continuation of the chain. The authors came to the conclusion about the correctness of this approach to the w(–O3) calculations in the work [14] when discussing other erroneous approaches [15–17] in our time to the calculations of the rate of the chain destruction of ozone in the lower stratosphere. When calculating the absolute values w(–O3), it was also taken into account that the concentration of component O in the Ox family is determined not only by the processes within the family itself but also when interacting with the components of the HOx, NOx, and ClOx families in which ozone is destroyed [14].

The altitudinal profiles of the partial rates of ozone destruction (thin lines) and the vertical profile of their total rate (curve in bold) in the HOx, NOx, ClOx, and Ox catalytic cycles for the conditions of June 1995 at latitude 50° N (see text) are calculated taking into account HCRs with the participation of particles of the Junge layer in the lower stratosphere. The inset compares the altitudinal profiles of the total ozone depletion rates with the participation of particles from the Junge layer (1) and without them (2).
The inset to Fig. 1 shows a comparison of the vertical profiles of the calculated values of the total rates of ozone destruction in the absence of HCRs (curve 1) and when they are taken into account (curve 2). From these data, it follows that the effect of sulfate aerosol on ozone depletion significantly changes depending on the altitude above the underlying surface, which is related to the variability of the concentrations of the components of these families considered above. Thus, at altitudes <16 km, the presence of sulfate particles inhibits the destruction of ozone. This is due to the effective capture of gas molecules N2O5 from the air by aerosol particles, which is due to their rapid hydrolysis (1) in the condensed phase. In this case, the competition between the processes of photodegradation of N2O5(g) with time τhν (N2O5(g) \(\xrightarrow{{h{{\nu }}}}\) NO3(g) + NO2(g)) and their capture by aerosol particles (τcapture). In this case, due to the high rate of reaction (1) of the hydrolysis of N2O5 [13], we can assume that the dynamics of this process is determined by the dynamics of their capture: τcapture = 4r/αϖL. Here α = 0.1 is the so-called coefficient of the reactive capture of N2O5 [13], and ϖ is the average thermal velocity of N2O5(g) molecules in cm/s. The estimates show that diffusion in a gas during the capture of N2O5(g) turns out to be a limiting stage only at r ≥ 10 microns. At altitudes up to 16 km, the capture of N2O5(g) in the considered competing processes turns out to be predominant. Its implementation is accompanied by a decrease in the concentrations of NO and NO2, as well as a decrease in the rate of ozone destruction in the nitrogen cycle (see Fig. 1). At the same time, although to a much lesser extent, the concentrations of OH and HO2 also decrease at these altitudes, which also leads to a decrease in the rates of ozone destruction in the HOx-loop. This negative effect of aerosol particles on the speed of the HOx and NOx cycles at these altitudes only partially compensates the increased rate of ozone destruction in the Ox and ClOx cycles.
As they rise above the underlying surface, the volume fraction of aerosol particles decreases, and the rate of capture of molecules N2O5 falls off, i.e., τcapture increases. In contrast, the photodissociation coefficient N2O5, \({{j}_{{{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}}}}\) = \(\tau _{{h{{\nu }}}}^{{ - {\text{1}}}}\) increases as it rises above the underlying surface, which leads to a decrease in the lifetime of N2O5 molecules and an increase in their photodegradation rate, i.e., τhν decreases. All this leads to an increase in the concentrations of NO and NO2(g), as well as an increase in the rate of ozone destruction not only in nitrogen, but also in other cycles (see Fig. 1). The volumetric content of aerosol particles turns out to be critical: Lcr ≥ 4r\({{j}_{{{{{\text{N}}}_{{\text{2}}}}{{{\text{O}}}_{{\text{5}}}}}}}\)/3αϖ ≥ 3 × 10–13. The given estimate Lcr is in agreement with the data of the calculations of the volumetric content of sulfate aerosol particles at the lower boundary of the Junge layer (10 km). Most of the N2O5(g) molecules for this reason passes into sulfate aerosol particles, which slows the destruction of ozone in the NOx and HOx cycles.
At altitude ranging from ≤17 to ≈23 km, the calculated w(–O3)HCR values turn out to be somewhat smaller (≈10–15%) in comparison with w(–O3). This means that, at these altitudes, the presence of aerosol particles in the lower stratosphere contributes to the acceleration of the ozone destruction process. In this case, the activation of the process of its destruction is provided mainly by an increase in the rate of the reactions of the HOx-cycle due to the suppression of the OH/HO2 + NO(NO2 processes maintained at these altitude), although a certain role is also played by the reactions of the ClOx-cycle (chlorine activation). The dynamics of ozone destruction is also affected by the increase in the rate of ozone destruction in the NOx-cycle as it rises, which masks the activation of this process in the HOx- and ClOx-loops. As noted, the increase in the rate of ozone destruction in the NOx-cycle is due to the increase in the concentrations of NO, NO2, and NO3 in the ascent due to the decrease in the speed of capturing N2O5 due to the decrease in the concentration of aerosol particles (see inset in Fig. 1).
At altitudes >22 km, the effect of sulfate aerosol particles on the rate of ozone destruction practically degenerates. This is related not only to the decrease in the volume fraction of aerosol particles and the increase in the content of sulfuric acid in them as they rise above the underlying surface but also with the increase in the rate of the gas-phase reactions. The decrease in the volume fraction of aerosol particles is due to the increase in temperature and decrease in the relative humidity of the air with an increase in the altitude above the underlying surface. As a consequence, sulfuric acid from the sulfate particles passes into the gas phase, which leads to the evaporation of the sulfate aerosol particles. The origin of the upper boundary of the Junge layer is also related to these processes [2, 18]. At the upper boundary of the latter, the volume fraction of the sulfate aerosol particles decreases approximately by a factor of three and the photodissociation coefficient increases approximately by a factor of 1.5. In such conditions, this leads to the fact that τcapture/τhν \( \gg \) 1 and most of the N2O5 molecules remain in the gas phase.
Figure 2 shows the height profiles of the relative contributions of the ClOx, Ox, HOx, and NOx cycles in the destruction of ozone in the summer at a latitude of 50° N. It can be seen that at altitudes <16 km, the contribution of ozone destruction reactions in the HOx-loop is dominant. When it rises to an altitude of 10 to 16 km, its share ranges from ∼98% to ∼60% of the entire ozone destroyed in the lower stratosphere, and the contribution of the NOx-cycle at altitudes <16 km is not more 10%. Moreover, in the Ox- and ClOx-cycles, in total, not more than 6% (∼2.2% + 3.7%) of ozone is destroyed. This differs markedly from the calculated distribution of the contributions of these cycles to ozone depletion when HCRs with the participation of particles of the Junge layer are ignored. Thus, the share of the Ox-, HOx-, ClOx-, and NOx-cycles comes to, respectively, ∼5%, ∼69%, ≤1%, and ∼25%. As noted, this is mainly due to the suppression at these altitudes of the reactions of ozone destruction in the NOx-cycle, which is caused by the effective capture of N2O5 molecules.

The altitudinal profiles of the relative contributions of the HOx, NOx, ClOx, and Ox catalytic cycles in ozone destruction, taking into account heterogeneous chemical reactions with the participation of particles of the Junge layer at latitude 50° N, are shown.
CONCLUSIONS
The paper presents the results of numerical calculations taking into account the effect of particles of the Junge layer on the rate of ozone destruction. The calculations indicate a significant effect of the capture of N2O5 from the air by these particles on the concentration of most of the components of the NOx-, HOx-, and ClOx-cycles of ozone destruction. At the same time, the considered changes in the concentrations of the components of these families and changes in the rates of ozone destruction are not directly related to the HCRs. These changes are due only to HCRs with the participation of N2O5/СlONO2 and particles of the Junge layer. Their effect is due to the decrease in the concentrations of the components of the NOx-family and the suppression of the reactions of their components with the components of the HOx and ClOx families. Therefore, the effect of the capture from the air of the N2O5 molecule needs to be taken into account even in the summer when calculating the dynamics of ozone destruction in the catalytic HOx-, NOx-, ClOx-, and Ox-cycles in the lower stratosphere.
REFERENCES
I. K. Larin, A. E. Aloyan, and A. N. Ermakov, Khim. Fiz. 36 (1), 90 (2017).
C. E. Junge, C. W. Chagnon, and J. E. Manson, J. Meteorol. 18, 81 (1961).
I. V. Kumpanenko, A. V. Roshchin, N. A. Ivanova, E. I. Zelenina, T. C. Volchenko, and E. O. Panin, Russ. J. Phys. Chem. B 12, 58 (2018).
V. V. Zelenov, E. V. Aparina, V. I. Kozlovskiy, I. V. Sulimenkov, and A. E. Nosyrev, Russ. J. Phys. Chem. B 12, 343 (2018).
A. E. Aloyan, A. N. Yermakov, and V. O. Arutyunyan, Russ. J. Phys. Chem. B 13, 214 (2019).
R. J. Salawitch, S. C. Wofsy, P. O. Wennberg, et al., Geophys. Res. Lett. 21, 2547 (1994).
I. K. Larin, A. E. Aloyan, and A. N. Ermakov, Russ. J. Phys. Chem. B 10, 860 (2016).
C. Voigt, H. Schlager, B. P. Luo, et al., Atmos. Chem. Phys. 5, 1371 (2005).
http://dataportal.ucar.edu/metadata/acd/software/Socrates/Socrates.thredds.xml.
G. Myhre, T. F. Berglen, C. L. E. Myhre, et al., Tellus 56B, 294 (2004).
D. R. Hanson, J. Phys. Chem. A 102, 4794 (1998).
Q. Shi, J. T. Jayne, C. E. Kolb, et al., J. Geophys. Res. 106, 24259 (2001).
I. K. Larin, Russ. J. Phys. Chem. B 11, 189 (2017).
G. Brasseur and S. Solomon, Aeronomy of the Middle Atmosphere: Chemistry and Physics of the Stratosphere and Mesosphere, 3rd ed. (Springer, Montreal, Canada, 2005).
D. J. Jacob, Introduction to Atmospheric Chemistry (Princeton Univ. Press, Princeton, 1999).
T. Shimazaki, Minor Constituents in the Middle Atmosphere (Terra Sci., Tokyo, Japan, 1985).
K. S. Carslaw, T. Peter, and S. L. Clegg, Rev. Geophys. 35, 125 (1997).
Funding
This study was supported by the Russian Foundation for Basic Research (project nos. 19-05-00080 and 19-05-50007 (Microworld)) and a state assignment of Semenov Federal Research Center for Chemical Physics, Russian Academy of Sciences (registration number AAAA-A20-120011390097-9).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Larin, I.K., Aloyan, A.E. & Ermakov, A.N. Influence of Particles of the Junge Layer on the Rate of Ozone Destruction in the Lower Stratosphere. Russ. J. Phys. Chem. B 15, 577–581 (2021). https://doi.org/10.1134/S199079312103009X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S199079312103009X