
Abstract—
Using random (combinatorial) DNA-libraries with various degrees of diversity, it was shown that their amplification by polymerase chain reaction in real time resulted in appearance of a maximum on amplification curves. The relative decrease of fluorescence after passing the maximum was directly proportional to the logarithm of the number of oligonucleotide sequence variants in the random DNA-library provided that this number was within the interval from 1 to 104 and remained practically unaltered when the number of variants was in the interval from 105 to 108. The obtained dependence was used in the course of the SELEX procedure to evaluate changes in the diversity of random DNA-libraries from round to round in selection of DNA-aptamers to the recombinant SMAD4 protein. As a result, oligonucleotides containing sequences able to form a site of SMAD4-DNA interactions known as SBE (SMAD-binding element) have been selected thus indicating that the SMAD4-SBE interaction dominates the aptamer selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Aptamers, considered as synthetic analogues of mAbs, are RNA or single-stranded DNA molecules that can fold with a formation of complex spatial structures and exhibit affinity for their molecular targets, comparable to that of mAbs [1, 2]. Selection of aptamers to a particular target (which could be low-molecular compounds, peptides, proteins, viruses, and cells) is performed in combinatorial RNA or DNA libraries by the SELEX method; as a rule, RNA libraries are obtained on the basis of DNA libraries [1, 3]. SELEX represents a series of repeated cycles (rounds), each of which includes binding of a random oligonucleotide library to the particular target, isolation of oligonucleotide/target complexes, amplification of the bound oligonucleotides by PCR, separation of DNA chains (in the case of DNA aptamer selection) to obtain a new random library [1, 2]. During selection, libraries are enriched with sequences exhibiting affinity for the target. Despite the apparent simplicity, the SELEX procedure is a complex process; its final result, the selection of high-affinity oligonucleotide sequences, is significantly influenced by various factors, such as a design of the initial combinatorial DNA library, the efficiency of separation of oligonucleotides bound and unbound to the target, optimization of PCR for amplification of oligonucleotides associated with the target, the choice of a ratio of the number of oligonucleotides and the target during selection, the number of selection rounds [3–5]. Usually, the last two parameters are selected empirically during aptamer selection based on the results of monitoring of changes in the oligonucleotide affinity to the target and the variety of a DNA library from round to round [4, 5].
The following methods are currently used to evaluate evolution of the combinatorial DNA library during selection that help to perform either qualitative or quantitative evaluation of the diversity of the library (the number of DNA oligonucleotide sequence variants): analysis of the denaturation (melting) and/or kinetics curves of DNA library renaturation after amplification procedures [6–8], nuclear magnetic resonance (to estimate the number of oligonucleotides with complex spatial conformation) [9], high-performance liquid chromatography under denaturing conditions [10], next generation sequencing [11, 12]. Qualitative assessment of the diversity of DNA libraries can be also performed using the form of amplification curves obtained by RT-PCR, which represent the fluorescence dependence for a dye intercalating into double-stranded DNA molecules (usually SYBR Green) on the number of amplification cycles [13]. While amplification curves for DNA fragments of the same nucleotide sequence are characterized by a sigmoid shape, the amplification of combinatorial DNA libraries passes through the fluorescence maximum followed by a decrease with subsequent plateau with a lower fluorescence value. This form of amplification curves is observed for combinatorial DNA libraries with both unmodified [13] and modified bases [14].
In this study, we have determined a quantitative relationship between the diversity of the combinatorial DNA library and such amplification curve parameter as a magnitude of the relative fluorescence decrease for the SYBR Green dye after passing through the maximum. The obtained dependence was used to monitor the selection of DNA aptamers to the human transcription factor SMAD4, which is considered as a potential target for targeted cancer therapies [15].
MATERIALS AND METHODS
Synthesis and Purification of DNA Oligonucleotides
The oligonucleotides shown in Table 1 were used in this study; these included: a combinatorial DNA library of 75 nucleotides (nt) in length with a random region of 32 nt long and flanking sites with constant sequences of 23 nt and 20 nt; a 75 nt oligonucleotide (oligo-C) with the sequences at the 5′- and 3′-ends identical to the flanking sequences of the DNA library; forward and reverse primers for amplification of the DNA library and oligo-C; a DNA aptamer (SBE-Apt) and a control oligonucleotide (SBE-Scr) biotinylated at the 3′-end. Oligonucleotides were synthesized on an ASM-800 DNA synthesizer (Biosset, Russia) in accordance with the manufacturer’s protocols. Synthesis of a random region of the DNA library was performed using a mixture of phosphoamidites in the ratio A : C : G : T = 1.5 : 1.25 : 1.15 : 1.0 [16]. The primers were purified by reverse phase chromatography, other oligonucleotides were purified by polyacrylamide gel electrophoresis (T = 12%, C = 5%) under denaturing conditions (7 M urea). Electrophoresis was performed in a TBE buffer. After staining with the SYBR Green I fluorescent dye (Life Technologies, USA) DNA was visualized by placing the gel on a UV transilluminator. The gel strip representing the upper part of the zone of the migrating material (it contained the most slowly migrated DNA molecules) was excised, cut into pieces, and DNA was eluted by incubating the gel pieces in deionized water. DNA was precipitated with ethanol in the presence of 0.3 M sodium acetate; the precipitate was dried and dissolved in deionized water. The concentration of the DNA library and primers was determined using a NanoDrop-1000 spectrophotometer (Thermo Fisher Scientific, USA) in accordance with the manufacturer’s instructions. The molecular mass of the oligonucleotides was calculated using the OligoCalculator program (http://mcb.berkeley.edu/labs/krantz/tools/ oligocalc.html), in the case of the random region of the DNA library, the average molecular mass of the nucleotide was assumed to be 325 Da.
Preparation and PCR Analysis of DNA Libraries with Varying Degrees of Diversity
An aliquot of the DNA library solution was sequentially diluted with deionized water to obtain a series of solutions with 10-fold different concentrations from 34 pM to 3.4 aM. 5 μL of each solution (corresponding to the amount of DNA ranged from 170 amol to 0.017 zmol) was used for RT-PCR performed using the qPCRmix-HS SYBR PCR mixture (Evrogen, Russia) and a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA). The volume of the PCR assay was 25 μL; the concentration of each primer was 400 nM. Each amplification cycle included 45 s incubations of the sample at 95°C, 56°C, and 72°C. The reaction was stopped after passing the maximum value of SYBR Green fluorescence. The contents of the PCR tube were used for preparative PCR (25 PCR samples of 100 μL each). 15 cycles of preparative PCR were performed on a Tetrad 2 thermocycler (Bio-Rad) using the qPCRmix-HS PCR mixture (Eurogen). The concentration of primers and the amplification conditions were the same as described above for RT-PCR. Preparative PCR products were concentrated using Vivaspin 2 centrifuge concentrators (Sartorius Stedium Biotech, Germany), and then purified by electrophoresis as described above. Ethanol precipitation of DNA was carried out in the presence of glycogen (Thermo Fisher Scientific). DNA concentration was determined on a NanoDrop 1000 spectrophotometer. The resultant DNA preparations were analyzed by RT-PCR. 170 amol of DNA was added to each PCR assay; RT-PCR conditions were the same as described above.
Expression and Purification of Recombinant SMAD4
Recombinant SMAD4 protein (rSMAD4), containing the N-terminal His6-tag was expressed in competent E. coli cells of the strain C41 transformed with plasmid pET23a carrying an insert encoding the SMAD4 protein. The cells of the transformed strain (further abbreviated as C41M) were sonicated and the cell lysate was cleared by centrifugation (4°C, 10 000 g, 40 min). rSMAD4 was purified using CL-4B Sepharose columns (Thermo Scientific) saturated with cobalt ions. The lysate was passed through the column, bound proteins were eluted with an imidazole gradient. The maximum elution of rSMAD4 was observed in the range of imidazole concentrations of 60−80 mM. Expression of rSMAD4 was confirmed by the results of electrophoretic (Fig. 1) and mass spectrometric (Supplementary materials, Fig. S1 and Table S1) analyses. Protein electrophoresis was performed according to the method of Laemmli [17]; the gels were stained with the Coomassie G-250 dye (Bio-Rad). Protein concentration was determined using the colorimetric method with bicinchoninic acid using the commercial Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).

The results of electrophoretic analysis of cell lysates of E. coli strain C41 (track 1) and C41M (track 2), as well as material bound to the column loaded with Sepharose CL-4B saturated cobalt ions and eluted in the imidazole concentration gradient range of 60−80 mM (track 3). Track 4 shows molecular mass markers (molecular mass values are shown on the right). The arrow indicates the position of the rSMAD4 band on the gel.
Selection of DNA Aptamers for SMAD4
For selection, rSMAD4 (molecular mass of 60.5 kDa) was immobilized on Dynabeads His-Tag Isolation & Pulldown (“Life Technologies”) magnetic beads. A commercial suspension of beads (10 μL), washed and equilibrated in 10 mM Tris-HCl buffer (pH 7.9), 500 mM NaCl, 0.02% Tween-20, was mixed with 150 μL of the protein solution (0.1 mg/mL) in the same buffer and incubated for 15 min; after the incubation the beads were washed and resuspended in 50 μL of SB buffer (10 mM Tris-HCl, pH 7.4, 500 mM NaCl). For counter-selection, the His-His-His-His-His-His (His6) peptide was immobilized on magnetic beads. A suspension of beads with His6 (SB, 50 μL) was added to the DNA library solution (SB, 1 mL) containing 100 pmol of oligonucleotides (≈2.4 μg), and incubated for 30 min at room temperature. Beads were collected on a magnetic separator and the DNA library solution was withdrawn. The resultant solution was mixed with beads bearing immobilized rSMAD4. After incubation for 30 min at room temperature, the beads were collected on a magnetic separator, the solution was removed, and the beads were washed twice with 200 μL of SB and resuspended in 50 μL of deionized water. The suspension of beads was heated for 3 min at 95°C and immediately placed on a magnetic separator. The resultant oligonucleotide solution was used for preparative PCR, which was carried out as described above with the following exceptions: a “heavy” reverse primer was used for amplification (Table 1, the primer conjugated to five hexaethylene glycol molecules at its 5'-end); the number of PCR samples ranged from 40 to 50, and the number of amplification cycles varied from 20 to 25. The amplicons were concentrated as described above. DNA strands were separated by electrophoresis due to the difference in their electrophoretic mobility (Supplementary materials, Fig. S2). The elution of the “light” chain from the gel, DNA precipitation, and determination of DNA concentration were performed as described above for DNA libraries. The resultant oligonucleotides were analyzed by RT-PCR, as described above, and then were used for the next round of selection.
After the last round of selection, PCR products were cloned using the Quick-TA kit (Eurogen) in accordance with the manufacturer’s recommendations and 10 clones were selected for sequencing which was performed by Eurogen.
Analysis of DNA Aptamer Binding to rSMAD4
The oligonucleotides SBE-Apt and SBE-Scr (Table 1), biotinylated at the 3'-end during their synthesis, were immobilized on streptavidin-bearing Dynabeads MyOne Streptavidin C1 magnetic beads (Thermo Scientific), following the manufacturer’s recommendations. The beads were mixed with the cleared cell lysate of the C41M strain containing expressed rSMAD4, incubated in the SB buffer for 30 min at room temperature under constant stirring, and isolated using a magnetic separator. The material bound to the magnetic beads was eluted by incubation in the electrophoresis sample buffer (containing 2% sodium dodecyl sulfate), after that the electrophoretic analysis of the eluate was performed as described above.
RESULTS AND DISCUSSION
PCR Analysis of DNA Libraries of Varying Degrees of Diversity
The DNA libraries used in the work were “degenerated” since the number of nucleotide sequence variants was less than theoretically possible [1, 3]. Indeed, the number of combinations of four types of nucleotides within a random region of the length of 32 nt is 432 ≈ 1.8 × 1019 variants, while 100 pmol (≈2.4 μg) of the DNA library used in the work contained only 6 × 1013 DNA molecules. Consequently, 170 amol will contain 108 DNA molecules, and 0.017 zmol will contain 10 DNA molecules, each of which will be represented by its sequence of the random region. The PCR amplification of these DNA oligonucleotides will increase their number many times without a significant change in the number of sequence variants, thus making it possible to obtain DNA libraries that differ in the variety of variants of nucleotide sequences from 108 to 101. Figure 2 shows the results of the analysis of such libraries by RT-PCR. While during amplification of DNA oligonucleotides with the same sequence (oligo-C) the shape of the amplification curve has the expected sigmoid form (Fig. 2a), amplification of DNA libraries is characterized by a maximum on amplification curves, and the relative decrease in fluorescence of the SYBR Green dye after reaching the maximum differs for libraries with a various degree of diversity (Fig. 2b).

Amplification curves presented as normalized fluorescence values of the SYBR Green intercalating dye for different amplification cycles. (a) Oligo-C; (b) DNA libraries of various degrees of diversity (the number of sequence variants): 1—108, 2—106, 3—104, 4—103, 5—102, 6—101. Normalization of the amplification curves was carried out by dividing the fluorescence values of the SYBR Green dye by its value at the 35th cycle for oligo-C (a) and at the maximum value of fluorescence for DNA libraries (b). The amount of DNA in the PCR sample was 170 amol. RT-PCR; each cycle included 45-s incubation at 95°C, 56°C, and 72°C; the concentration of primers was 400 nM.
For a quantitative measure of the decrease in fluorescence we have used the value ΔFr = (Fm – F35)/Fm, where Fm and F35 are the maximum fluorescence values on the amplification curve and the fluorescence values at the 35th RT-PCR cycle, respectively. Figure 3 shows insignificant changes in the ΔFr values when the diversity of the library decreases from 108 to 105 sequence variants. However, upon a further decline in diversity, ΔFr begins to decrease rapidly. If for diversity 1 (DNA oligonucleotides with the same sequence) the ΔFr value is taken equal to zero (Fig. 2a), then in the interval of 1–104 sequence variants, the dependence of ΔFr on the degree of diversity can be well approximated by the following relation: ΔFr = 0.126 log N, where N is the expected number of sequence variants in the DNA library (Fig. 3; the linear dependence shown in the interval of 1–104 was obtained by linear regression, the Pearson correlation coefficient was 0.987). We cannot rule out that the obtained value of the coefficient of proportionality is applicable only to this DNA library (75 nt long, with a random region of 32 nt) and will be different when the ratio between the length of the random region and the total length of the flanking sequences changes.

A relative decrease of SYBR Green dye fluorescence calculated as ΔFr = (Fm – F35)/Fm, where Fm and F35 are the maximum fluorescence values on the amplification curve and the fluorescence values at the 35th RT-PCR cycle as a function of diversity (expected number of sequence variants) of DNA libraries. The straight line shown for the number of sequence variants from 100 to 104 was obtained by linear regression (Pearson correlation coefficient r = 0.987); the dependence is described by the equation ΔFr = 0.126 log N, where N is the expected number of sequence variants in the DNA library. All measurements were performed in triplicates. Mean values and standard deviations are shown for triplicates.
It is possible that the unusual shape of amplification curves in the case of DNA libraries may be attributed to the fact that all, or a significant part, of the bases in the region with a random sequence are unpaired. Indeed, the fluorescence measurement in PCR samples were performed at the end of the primer elongation step (at 72°C), when in the case of DNA oligonucleotides with the same sequence they are all in the form of fully base-paired DNA duplexes. As the number of complementary oligonucleotides increases during amplification, they begin to compete with primers, thus slowing and finally completely inhibiting amplification. However, in this case fragments of duplex DNA with completely complementary bases are formed thus resulting in the amplification curve of the classical sigmoid shape (Fig. 2a). In the case of DNA libraries, flanking regions of oligonucleotides begin to compete with primers, while random regions of the resulting duplex DNAs are non-complementary and represent single-stranded regions. Thus, starting from a certain amplification cycle, the number of fully duplex DNA molecules obtained due to primer elongation (and, correspondingly, the number of SYBR Green molecules intercalating between base pairs) begins to decrease; this leads to a decrease in the fluorescence level and the appearance of a maximum on the amplification curves (Fig. 2b). Obviously, as the diversity of the DNA library decreases, the probability of the formation of fully complementary DNA duplexes increases, and the maximum on the amplification curves becomes less pronounced (Figs. 2, 3).
Selection of DNA Aptamers to rSMAD4
SMAD4 is a common partner for proteins of the R‑SMAD group involved in formation of transcriptionally active complexes induced by transforming growth factor beta (TGF-β) [15]. SMAD4 consists of two domains, MH1 and MH2; MH1 binds to genomic DNA, while MH2 binds to one of R-SMAD proteins [18]. The MH1 domain specifically binds to the duplex formed by the 5'-GTCT-3' sequence and its 5'-AGAC-3' complement, known as SBE (SMAD-binding element) [18, 19].
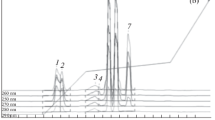
The selection of DNA aptamers for rSMAD4 was controlled by evaluating the diversity of DNA libraries from round to round. Figure 4 shows that the maximum on the amplification curves becomes less pronounced as the number of selection rounds increases. A comparison of results given in Figs. 3 and 5 shows that after the first two rounds of selection, the diversity of the library remains relatively high: at least 106 variants. After the third round of selection, the variety decreases to approximately 104 variants, and after the fourth, it is in the range of 103−104 variants and basically remains unchanged after the fifth round. At this stage, the SELEX procedure has been changed: the number of beads with rSMAD4 added to the DNA library was reduced by one order. As can be seen from Figs. 4 and 5, this significantly changed the diversity of the library after the sixth round: it ranged between 102–103 sequence variants (Figs. 3 and 5). After the eighth round of selection, the diversity became no more than 100 sequence variants (Figs. 3 and 5), and at this stage we decided to clone and sequence DNA fragments in order to analyze the sequences presented in the DNA library. The analysis has shown that among ten sequenced clons of PCR products (Table 2), two included the sequences 5'-GTCT-3' and 5'-AGAC-3' and were able to form a hairpin structure; the duplex part of this hairpin included SBE, and one sequence contained two sequences of 5'-GTCT-3' and 5'-AGAC-3', which could form two SBEs in the duplex part of the DNA hairpin (sequence no. 9, Table 2). A schematic representation of the three-dimensional structure formed by this sequence is shown in Fig. 6 (the structure was obtained using the Mfold program [20]).

Amplification curves (normalized fluorescence values of the intercalating dye SYBR Green at different amplification cycles) for DNA libraries obtained after rounds of aptamer selection for rSMAD4. The numbers indicate the selection rounds.

Relative decrease in fluorescence (ΔFr = (Fm – F35)/Fm, as in Fig. 2) for DNA libraries obtained in different rounds of aptamer selection for rSMAD4. All measurements were performed in triplicates. Mean values and standard deviations are shown for triplicates.

The structure formed by oligonucleotide no. 9 from Table 2. The structure was obtained using the Mfold program (http://unafold.rna.albany.edu/?q=mfold/DNA-Folding-Form). Bold lines indicate the SBE position.
The results were used for construction of the DNA aptamer (SBE-Apt, Table 1). The SBE-Apt aptamer was able to bind selectively rSMAD4 in E. coli lysates (Fig. 7). At the same time, the SBE-Scr oligonucleotide able to form the identical DNA hairpin but without the SBE formation due to the fact that the nucleotides in the corresponding regions were randomly mixed did not show significant interaction with rSMAD4 (Fig. 7). It is likely that the site of interaction of the rSMAD4 protein MH1 domain with SBE is the dominant DNA binding site that determines the selection in the DNA libraries of oligonucleotides forming hairpin structures with a duplex part containing SBEs.

Results of electrophoretic analysis of rSMAD4 binding to the SBE-Apt DNA aptamer immobilized on magnetic microparticles. M—molecular mass markers (indicated on the left); 1—microparticles with immobilized SBE-Apt; 2—beads with an immobilized control SBE-Scr oligonucleotide (Table 1); 3—beads without immobilized oligonucleotides. The arrow indicates the rSMAD4 band position on the gel. Lower bold bands are streptavidin monomers.
CONCLUSIONS
Using a combinatorial DNA library of 75 nt long with a random region of 32 nt long as an example, we have demonstrated that the relative decrease in fluorescence after passing the maximum is directly proportional to the logarithm of the number of variants of oligonucleotide sequences in the range from 1 to 104 and remains basically unchanged provided that the number of variants varies in the range from 105 up to 108. The obtained dependence was used in the SELEX procedure to evaluate changes in the diversity of random DNA libraries from round to round during the selection of DNA aptamers to the recombinant protein SMAD4. The described procedure resulted in the selection of oligonucleotides containing sequences that form the site of DNA interaction with SMAD4, known as SBE.
REFERENCES
Radko, S.P., Rakhmetova S.Yu., Bodoev N.V., and Archakov A.I. Biochemistry (Moscow)Suppl. Ser. B: Biomed. Chem., 2007, vol. 1, pp. 198−209. https://doi.org/10.1134/S1990750807030043
Zhang, Y., Lai, B.S., and Juhas, M., Molecules, 2019, vol. 24, E941. https://doi.org/10.3390/molecules24050941
Komarova, N. and Kuznetsov, A., Molecules, 2019, vol. 24, E3598. https://doi.org/10.3390/molecules24193598
Wang, T., Chen, C., Larcher, L.M., Barrero, R.A., and Veedu, R.N., Biotechnol. Adv., 2019, vol. 37, pp. 28−50. https://doi.org/10.1016/j.biotechadv.2018.11.001
Mencin, N., Smuc, T., Vranicar, M., Mavri, J., Hren, M., Galesa, K., Krkoc, P., Ulrich, H., and Solar, B., J. Pharm. Biomed. Anal., 2014, vol. 91, pp. 151−159. https://doi.org/10.1016/j.jpba.2013.12.031
Vanbrabant, J., Leirs, K., Vanschoenbeek, K., Lammertyn, J., and Michiels, L., Analyst, 2014, vol. 139, pp. 589−595. https://doi.org/10.1039/c3an01884a
Charlton, J. and Smith, D., RNA, 1999, vol. 5, pp. 1326−1332. https://doi.org/10.1017/s1355838299991021
Schutze, T., Arndt, P.F., Menger, M., Wochner, A., Vingron, M., Erdmann, V.A., Lehrach, H., Kaps, C., and Glokler, J., Nucleic Acids Res., 2010, vol. 38, e23. https://doi.org/10.1093/nar/gkp1108
Amano, R., Aoki, K., Miyakawa, S., Nakamura, Y., Kozu, T., Kawai, G., and Sakamoto, T., Sci. Rep., 2017, vol. 7, 283. https://doi.org/10.1038/s41598-017-00273-x
Muller, J., El-Maarri, O., Oldenburg, J., Potzsch, B., and Mayer, G., Anal. Bioanal. Chem., 2008, vol. 390, pp. 1033−1037. https://doi.org/10.1007/s00216-007-1699-8
Schutze, T., Wilhelm, B., Greiner, N., Braun, H., Peter, F., Morl, M., Erdmann, V.A., Lehrach, H., Konthur, Z., Menger, M., Arndt, P.F., and Glokler, J., PLoS One, 2011, vol. 6, e29604. https://doi.org/10.1371/journal.pone.0029604
Gu, G., Wang, T., Yang, Y., Xu, X., and Wang, J., PLoS One, 2013, vol. 8, e76109. https://doi.org/10.1371/journal.pone.0076109
Luo, Z., He, L., Wang, J., Fang, X., and Zhang, L., Analyst, 2017, vol. 142, pp. 3136−3139. https://doi.org/10.1039/c7an01131h
Lapa, S.A., Pavlov, A.S., Kuznetsova, V.E., Shershov, V.E., Spitsyn, M.A., Guseinov, T.O., Radko, S.P., Zasedatelev, A.S., Lisitsa, A.V., and Chudinov, A.V., Mol. Biol. (Moscow), 2019, vol. 53, pp. 460−469. https://doi.org/10.1134/S0026893319030099
Ullah, I., Sun, W., Tang, L., and Feng, J., J. Cancer, 2018, vol. 9, pp. 4018−4028. https://doi.org/10.7150/jca.20906
Lapa, S.A., Romashova, K.S., Spitsyn, M.A., Shershov, V.E., Kuznetsova, V.E., Guseinov, T.O., Zasedatelev, A.S., Radko, S.P., Timofeev, E.N., Lisitsa, A.V., and Chudinov, A.V., Mol. Biol. (Moscow), 2018, vol. 52, pp. 854−864. https://doi.org/10.1134/S0026893318060110
Laemmli, U.K., Nature, 1970, vol. 227, pp. 680−685. https://doi.org/10.1038/227680a0
Zawel, L., Dai, J.L., Buckhaults, P., Zhou, S., Kinzler, K.W., Vogelstein, B., and Kern, S.E., Mol. Cell, 1998, vol. 1, pp. 611−617. https://doi.org/10.1016/s1097-2765(00)80061-1
Shi, Y., Wang, Y.F., Jayaraman, L., Yang, H., Massague, J., and Pavletich, N.P., Cell, 1998, vol. 94, pp. 585−594. https://doi.org/10.1016/s0092-8674(00)81600-1
Zuker, M., Nucleic Acids Res., 2003, vol. 31, pp. 3406−3415. https://doi.org/10.1093/nar/gkg595
ACKNOWLEDGMENTS
The experiments were performed using the equipment available in the Center of the Collective Use the “Human Proteome” at the Institute of Biomedical Chemistry (IBMC). The authors are grateful to Dr. A.A. Moysa (Laboratory of Systems Biology, IBMC) for the synthesis and purification of the His6 peptide.
Funding
This work was financially supported by the Ministry of Education and Science of the Russian Federation (agreement no. 14.576.21.0096, the project unique identifier RFMEFI57617X0096).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
COMPLIANCE WITH ETHICAL STANDARDS
This article does not contain any research using humans or using animals as objects.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary materials are available in the electronic version of the article at the journal website (pbmc.ibmc.msk.ru).
Additional information
Translated by A. Medvedev
Abbreviations used: mAB—monoclonal antibodies; SBE—SMAD-binding element; SELEX—Systematic Evolution of Ligands by Exponential enrichment; PCR—polymerase chain reaction; RT-PCR—real-time polymerase chain reaction.
Rights and permissions
About this article

Cite this article
Radko, S.P., Lapa, S.A., Chudinov, A.V. et al. Evaluation of the Diversity of Random DNA-Libraries by the Shape of Amplification Curves for Estimation of the Efficiency of Aptamer Selection. Biochem. Moscow Suppl. Ser. B 14, 159–167 (2020). https://doi.org/10.1134/S1990750820020109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990750820020109