
Abstract
This work is aimed to develop several cationic amphiphiles based on amino acid derivatives of diethanolamine as potentially membrane-active antibacterial agents. The developed compounds contain two amino acid residues in the polar block and aliphatic chains of various length in the hydrophobic domain. Amphiphiles were obtained in preparative amounts sufficient to confirm their structures and perform a study of antibacterial activity. The synthesized samples based on β-Ala (4c) and GABA (4d) with aliphatic C12 chain in the hydrophobic domain showed a promising level of antimicrobial activity against gram-positive (B. subtilis) and gram-negative (E. coli) bacteria (minimal inhibitory concentration, MIC, 1 μg/mL). Amphiphiles containing aromatic amino acids L-Phe (6a) and L-Trp (6b) in the polar head group and C8 hydrocarbon chain exhibited an antibacterial activity against B. subtilis with MIC of 1 μg/mL. The obtained data on antimicrobial activity make the selected compounds attractive for further detailed study of their mechanism of action.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The rapid spread of bacteria resistant to existing antibiotics poses many challenges to the health care system. Antibiotics that are part of standard treatment protocols eventually become ineffective against multidrug-resistant bacteria, resulting in a severe course of infectious diseases. Many research groups around the world are searching for effective means to combat pathogens that can complement the antibacterial therapeutic arsenal.
Antimicrobial peptides (AMPs) are believed to be a new type of possible antimicrobial agents of natural origin [1]. They combine antimicrobial, angiogenic, and anti-inflammatory activity with immunomodulatory effects [2]. However, the high cost of consumables, insufficient study of physicochemical and biological properties, instability of the obtained product or its toxicity to the host’s own cells make it difficult to widely use such biologically active structures [3].
Disadvantages of AMPs stimulated the development of new membrane-active substances, peptidomimetics [4]. These are synthetic derivatives of amino acids and peptides designed to mimic the basic functions and properties of the original pharmacophores. The choice of the membrane as a target provides an advantage of peptide agents over conventional antibiotics since the development of resistance to the peptidomimetics is slow or even absent. Such molecules retain their activity profile and selectivity of action, while they have greater bioavailability and stability under physiological conditions compared to classical AMPs. Over the past decade, a number of such agents have been synthesized, isolated, and studied [4]. Some of them are already used in the clinic [5] and some are undergoing clinical trials [6, 7].
One of the most promising trends is the use of cationic amphiphiles as peptidomimetics that have a simple design and high stability and show promising results of antimicrobial action [8]. The particular interest in these compounds stems from efficient, low-cost, and time-saving synthetic strategies. [9]. Main methods for the preparation of amphiphiles involve classical methods of peptide and lipid chemistry, which allows easy replacement or introduction of alternative fragments representing various amino acid analogs or aliphatic links [8].
Biological activity of amphiphiles largely depends on the architecture of the synthesized molecules. It is not uniform, unlike prototypes consisting exclusively of α-amino acid residues but involves varying the structure of the main fragments. In addition, it is possible to synthesize small molecules whose central link is not represented by an amide backbone [10, 11]. In general terms, the structure of amphiphilic molecules is represented by one or two aliphatic chains, amino acid sites as a hydrophilic head group, and a spacer connecting these two domains. Varying the structural elements of the amphiphile can affect the bioavailability of the molecule and its toxic effect [12]. Studies show that there is a relationship between the length of the hydrophobic domain of the amphiphilic compound and the minimum inhibitory concentration (MIC) that is required to inhibit the growth of microorganisms [13].
The most important role in the positive dynamics of antibacterial action is played by the adjusting of the amphiphilicity of these compounds, in particular, the ratio of the hydrophilic part to the hydrophobic part, the hydrophilic-lipophilic balance (HLB) [14, 15]. The choice of optimal design of peptidomimetics determines their antimicrobial activity and reduces toxic effects.
The aim of this study is to develop a scheme of the preparation and synthesis of a series of novel antimicrobial peptidomimetics and to perform preliminary biological tests of their antibacterial activity. Cationic amphiphiles based on aliphatic derivatives of diethanolamine containing two amino acid residues in the polar block and differing in the length of the aliphatic chains in the hydrophobic domain were chosen as target compounds. Published data on molecules with a similar structure [8, 16, 17] suggest that the newly synthesised molecules may be active against bacterial cell membranes.
MATERIALS AND METHODS
Materials. Reagents for the synthesis were commercially available and used without pre-treatment. 1H-NMR spectra were recorded in deuterated solvent on a BrukerWM-300 pulsed NMR spectrometer (Germany) with an operating frequency of 300 MHz. The internal standard was hexamethyldisiloxane. IR spectra of the substances were recorded on a Bruker EQUINOX 55 FTIR spectrometer (Germany). Mass spectra were recorded on a VISION 2000 time-of-flight mass spectrometer (UK) using the MALDI method; 2,4-dihydroxybenzoic acid (DHB) was used as a matrix. Thin-layer chromatography (TLC) was performed on Sorbfil (LLC Imide, Krasnodar, Russia) and Silufol plates (Czech Republic); preparative thin-layer chromatography was performed on TLC standard grade silica gel (Sigma-Aldrich, Germany). Column chromatography was performed on 0.040–0.063 mm silica gel (Merck, Germany). Substances were detected by thin-layer chromatography by heating over the flame of an alcohol burner or iodine vapor. Substances containing amino groups were detected in a 5% solution of ninhydrin followed by heating to 50°C. Aromatic compounds were detected under UV light on Alugram Xtra SIL G/UV254 plates.
Methods. Synthesis of cationic amphiphiles was performed by standard methods of peptide and lipid chemistry.
N-Octyldiethanolamine (2a). A mixture of 1 g (9.51 mmol) diethanolamine, 1.65 g (8.56 mmol) 1‑bromoctane, and 6.58 g (47.6 mmol) potassium carbonate in 20 mL acetonitrile was stirred at 80°C for 24 h. After completion of the reaction, the excess potassium carbonate was filtered off and the solvent was removed under vacuum. The reaction product was dissolved in 25 mL ethyl acetate, washed with distilled water (3 × 20 mL) and dried over anhydrous sodium sulfate. The product was isolated by column chromatography in a chloroform : methanol (9 : 1 v/v) system; 837 mg of product 2a (45%) was obtained.
1H NMR spectrum (DMSO-d6, δ, m.d.): 0.88 (t, 3H, CH3), 1.27 (s, 10H, CH2CH2(CH2)5CH3), 1.38 (p, 2H, CH2CH2(CH2)5CH3), 2.43 (t, 2H, CH2CH2(CH2)5CH3), 2.51 (t, 4H, CH2CH2NH), 3.40 (t, 4H, CH2CH2NH), 4.33 (s, 2H, OH).
N-Dodecyldiethanolamine (2b). N-Dodecyldiethanolamine was prepared similarly. From 1 g (9.51 mmol) of diethanolamine and 2.13 g (8.56 mmol) of 1-bromodododecane 1.7 g of product 2b (74%) were obtained.
1H NMR spectrum (DMSO-d6, δ, m.d.): 0.85 (t, 3H, CH3), 1.24 (s, 18H, CH2CH2(CH2)9CH3), 1.36 (p, 2H, CH2CH2(CH2)9CH3), 2.43 (t, 2H, CH2CH2(CH2)9CH3), 2.51 (t, 4H, CH2CH2NH), 3.40 (t, 4H, CH2CH2NH), 4.37 (s, 2H, OH).
O,O'-di-(N-(tert-butoxycarbonyl-β-alanyl)octyl)di-ethanolamine (3a). 225 mg (1.84 mmol) of 4-dimethylaminopyridine (DMAP) and a solution of 380 mg (1.84 mmol) of dicyclohexylcarbodiimide (DCC) in 5 mL of methylene chloride was added to a solution of 220 mg (1.15 mmol) of Boc-(β-Ala)-OH in 5 mL of anhydrous methylene chloride under stirring. The mixture was stirred at 0°C for 15 min. 100 mg (0.461 mmol) of product 2a in 5 mL methylene chloride was added to the reaction mass. The mixture was kept under vigorous stirring for 24 h. After completion of the reaction the precipitate of dicyclohexylurea was filtered off, the reaction mixture was dissolved in 50 mL ethyl acetate and washed with distilled water (3 × 50 mL) and dried over Na2SO4. The solvent was removed under vacuum, and the product was isolated by column chromatography in toluene : ethyl acetate (2 : 1 v/v) system. The yield of product 3a was 102 mg (40%).
1H NMR spectrum (CDCl3, δ, m.d.): 0.89 (t, 3H, CH3), 1.28 (s, 10H, CH2CH2(CH2)5CH3), 1.46 (s, 18H, CCH3), 1.94 (m, 2H, CH2CH2(CH2)5CH3), 2.53 (t, 4H, β-Ala: αCH2), 2.64 (t, 2H, CH2CH2(CH2)5CH3), 2.78 (t, 4H, CH2CH2NH), 3.41 (q, 4H, β-Ala: βCH2), 4.18 (t, 4H, CH2CH2NH), 5.25 (s, 2H, NH).
IR spectrum: (KBr), ν/cm–1: 3415 (N–H); 2949, 2894 (C–H); 1743 (C=O ester), 1720 (C=O, amide I); 1527 (N–C, amide II).
O,O'-di-(N-(tert-butoxycarbonyl-GABA)octyl)di-ethanolamine (3b). The reaction was performed in a similar manner. From 234 mg (1.15 mmol) of Boc-(GABA)-OH and 0.1 g (0.461 mmol) of compound 2a, 112 mg of product 3b (41%) was obtained.
1H NMR spectrum (CDCl3, δ, m.d.): 0.89 (t, 3H, CH3), 1.28 (s, 10H, CH2CH2(CH2)5CH3), 1.46 (s, 18H, CCH3), 1.82 (p, 4H, GABA: βCH2), 1.95 (m, 2H, CH2CH2(CH2)5CH3), 2.37 (t, 4H, GABA: αCH2), 2.52 (t, 2H, CH2CH2(CH2)5CH3), 2.78 (t, 4H, CH2CH2NH), 3.17 (m, 4H, GABA: γCH2), 4.15 (t, 4H, CH2CH2NH), 4.79 (s, 2H, NH).
IR spectrum: (KBr), ν/cm–1: 3386 (N–H); 2927, 2856 cm–1 (C–H); 1737 (C=O ester); 1677 (C=O, amide I); 1521 (N–C, amide II).
O,O'-di-(N-(tert-butoxycarbonyl-β-Ala)dodecyl)di-ethanolamine (3c). Compound 3c was prepared in a similar manner. From 0.34 g (1.8 mmol) of Boc-(β-Ala)-OH and 0.2 g (0.73 mmol) of compound 2b, 277 mg of product 3c (62%) was obtained.
1H NMR spectrum (CDCl3, δ, m.d.): 0.86 (t, 3H, CH3), 1.28 (s, 18H, CH2CH2(CH2)9CH3), 1.46 (s, 18H, CCH3), 1.63 (p, 2H, CH2CH2(CH2)9CH3), 2.53 (t, 4H, β-Ala: αCH2), 2.59 (t, 2H, CH2CH2(CH2)9CH3), 2.83 (t, 4H, CH2CH2NH), 3.37 (m, 4H, β-Ala: βCH2), 4.21 (t, 4H, CH2CH2NH), 5.22 (s, 2 H, NH).
IR spectrum: (KBr), ν/cm–1: 3311 (N–H); 2958, 2902 (C–H); 1739 (C=O ester); 1715 (C=O, amide I); 1519 (N–C, amide II).
O,O'-di-(N-(tert-butoxycarbonyl-GABA)dodecyl)di-ethanolamine (3d). Compound 3d was prepared in a similar manner. From 0.37 g (1.8 mmol) of Boc-(GABA)-OH and 0.2 g (0.73 mmol) of compound 2b, 280 mg of product 3d (60%) was obtained.
1H NMR spectrum (CDCl3, δ, m.d.): 0.87 (t, 3H, CH3), 1.24 (s, 18H, CH2CH2(CH2)9CH3), 1.46 (s, 18H, CCH3), 1.63 (p, 2H, CH2CH2(CH2)9CH3), 1.80 (m, 4H, GABA: βCH2), 2.36 (t, 4H, GABA: αCH2), 2.49 (t, 2H, CH2CH2(CH2)9CH3), 2.73 (t, 4H, CH2CH2NH), 3.14 (m, 4H, GABA: γCH2), 4.11 (t, 4H, CH2CH2NH), 4.75 (s, 2H, NH).
IR spectrum: (KBr), ν/cm–1: 3340 (N–H); 2914, 2855 cm–1 (C–H); 1735 (C=O ester); 1625 (C=O ester, amide I); 1521 (N–C, amide II).
O,O'-di-(β-alaniloctyl)diethanolamine bistrifluoroacetate (4a). 33 mg (0.3 mmol) of trifluoroacetic acid was added to a solution of 11 mg (0.02 mmol) of substance 3a in 5 mL anhydrous methylene chloride. The mixture was stirred for 2 h at 0°C. After completion of the reaction, the solvent and excess trifluoroacetic acid were distilled off at a rotary evaporator. The final oily product 4a was obtained in quantitative yield.
Mass spectrum, m/z: 382.18 [M + Na]+; 398.20 [M + K]+.
O,O'-di-(GABA-octyl)diethanolamine bistrifluoroacetate (4b). Compound 4b was prepared in a similar manner. From 14 mg (0.024 mmol) of compound 3b and 41 mg (0.36 mmol) of trifluoroacetic acid, product 4b was obtained in quantitative yield.
Mass spectrum, m/z: 410.12 [M + Na]+; 426.18 [M + K]+.
O,O'-di-(β-alanyl dodecyl)diethanolamine bistrifluoroacetate (4c). Compound 4c was prepared in a similar manner. From 12 mg (0.02 mmol) of compound 3c and 41 mg (0.36 mmol) of trifluoroacetic acid, product 4c was obtained in quantitative yield.
Mass spectrum, m/z: 438.213 [M + Na]+; 454.209 [M + K]+.
O,O'-di-(GABA-dodecyl)diethanolamine bistrifluoroacetate (4d). Compound 4d was prepared in a similar manner. From 10 mg (0.016 mmol) of compound 3d and 27 mg (0.23 mmol) of trifluoroacetic acid, product 4d was obtained in quantitative yield.
Mass spectrum, m/z: 466.29 [M + Na]+; 485.31 [M + K]+.
O,O'-di-(N-(tert-butoxycarbonyl-L-Phe)octyl)di-ethanolamine (5a). The reaction to obtain 5a was carried out similarly to the reaction for compound 3a. From 0.1 g (0.46 mmol) of 2a and 0.427 g (1.6 mmol) of Boc-(Phe)-OH, 0.24 g of product 5a (72%) was obtained.
1H NMR spectrum: (CDCl3, δ, m.d.): 0.88 (t, 3H, CH3), 1.27 (s, 10H, CH2CH2(CH2)5CH3), 1.41 (s, 18H, CCH3), 1.54 (m, 2H, CH2CH2(CH2)5CH3), 2.46 (t, 2H, CH2CH2(CH2)5CH3), 2.67 (t, 4H, CH2CH2NH), 3.10 (m, 4H, NHCHCH2C6H5), 4.12 (t, 4H, CH2CH2NH), 4.56 (m, 2H, NHCHCH2C6H5), 5.02 (s, 2H, NH), 7.14–7.32 (m, 10H, C6H5).
IR spectrum: (KBr), ν/cm–1: 3443 (N–H); 3070, 3057, 750, 709 (arC–H); 2982, 2851 cm–1 (C–H); 1751 (C=O); 1678 (C=O ester, amide I); 1516 (N–C, amide II).
O,O'-di-(N-(tert-butoxycarbonyl-L-Trp)octyl)di-ethanolamine (5b). The reaction to obtain 5b was carried out similarly to compound 3a. From 0.1 g (0.4 mmol) of 2a and 0.490 g (1.6 mmol) of Boc-(Trp)-OH, 0.2 g of product 5b (56%) was obtained.
1H NMR spectrum (CDCl3, δ, m.d.): 0.90 (t, 3H, CH3), 1.25 (s, 10H, CH2CH2(CH2)5CH3), 1.48 (s, 18H, CCH3), 1.57 (m, 2H, CH2CH2(CH2)5CH3), 2.24 (t, 2H, CH2CH2(CH2)5CH3), 2.35 (t, 4H, CH2CH2NH), 3.28 (m, 4H, NHCHCH2C8H5NH), 3.94 (t, 4H, CH2CH2NH), 4.62 (m, 2H, NHCHCH2C6H5NH), 5.21 (s, 2H, NH), 7.00–7.58 (m, 12H, C8H5NH), 8.68 (m, 2H, C8H5NH).
IR spectrum: (KBr), ν/cm–1: 3361 (N–H); 3063, 3030, 749, 701 (arC–H); 2928, 2855 cm–1 (C–H); 1721 (C=O ester); 1715 (C=O ester, amide I); 1500 (N–C, amide II).
O,O'-di-(N-(tert-butoxycarbonyl-L-Tyr)octyl)di-ethanolamine (5c). The reaction for the preparation of 5c was carried out in a similar way. From 0.1 g (0.4 mmol) of 2a and 0.405 g (1.4 mmol) of Boc-(Tyr)-OH, 96 mg of product 5c (28%) was obtained.
1H NMR spectrum (CDCl3, δ, m.d.): 0.88 (t, 3H, CH3), 1.26 (s, 10H, CH2CH2(CH2)5CH3), 1.43 (s, 18H, CCH3), 1.58 (m, 2H, CH2CH2(CH2)5CH3), 2.34 (t, 2H, CH2CH2(CH2)5CH3), 2.73 (t, 4H, CH2CH2NH), 2.94 (m, 4H, NHCHCH2C6H4), 3.65 (t, H, CH2CH2NH), 4.53 (m, 2H, NHCHCH2C6H4), 4.99 (s, 2H, NH), 6.75–7.04 (m, 10H, C6H5), 7.25 (s, 2H, OH).
IR spectrum: (KBr), ν/cm–1: 3474 (N–H); 3072, 3061 (arC–H); 2934, 2854 cm–1 (C–H); 1692 (C=O); 1643 (C=O ester, amide I); 1514 (N–C, amide II).
O,O'-di-(L-Phe-octyl) diethanolamine bistrifluoroacetate (6a). 29 mg (0.25 mmol) of trifluoroacetic acid was added to a solution of 12 mg (0.017 mmol) of substance 5a in 7 mL anhydrous CH2Cl2. The mixture was stirred for 3 h at 0°C. After completion of the reaction, the solvent and excess trifluoroacetic acid were distilled off at a rotary evaporator. The final oily product 6a was obtained in quantitative yield.
Mass spectrum, m/z: 534.24 [M + Na]+; 550.22 [M + K]+.
O,O'-di-(L-Trp-octyl)diethanolamine bistrifluoroacetate (6b). Compound 6b was prepared in a similar manner. From 12 mg (0.013 mmol) of compound 5b and 20 mg (0.20 mmol) of trifluoroacetic acid, product 6b was obtained in quantitative yield.
Mass spectrum, m/z: 612.16 [M + Na]+; 628.22 [M + K]+.
O,O'-di-(L-Tyr-octyl)diethanolamine bistrifluoroacetate (6c). Compound 6c was prepared in a similar manner. From 12 mg (0.016 mmol) of compound 5b and 28 mg (0.24 mmol) of trifluoroacetic acid, product 6c was obtained in quantitative yield.
Mass spectrum, m/z: 566.40 [M + Na]+; 581.37 [M + K]+.
Determination of the minimum inhibitory concentration (MIC) by the agar diffusion method. Study of the antibacterial activity of the obtained amphiphiles was carried out in collaboration with the Basic department of the FSBI Gause Institute of New Antibiotics. Prepared suspensions of Bacillus subtilis 534 and Escherichia coli M17 with a concentration of 1.5 × 108 CFU/mL and an optical density of 0.5 McFarland units were used as test microorganisms. Aliquots (100 μL) of the suspension were applied to Petri dishes and filled with 10 mL of warm nutrient agar, stirred, and left to harden. Then sterile discs of thick filter paper (6 pieces) of equal size and weight, with an inner diameter of 6.0 ± 0.1 mm, were placed at equal distances from each other on the surface of the agar in the dishes with cultures. Aliquots of the test and control samples were applied to the discs: 1, 10 and 100 µL each, corresponding to 1, 10 and 100 µg/mL of the substance. The plates were then incubated at room temperature for 1–2 h, followed by incubation at 36 ± 1°C for 16–18 h. At the end of the experiment, the diameter of the growth suppression zones of the test microorganism was determined with an accuracy of 1 mm. The experiment was performed three times. Ampicillin (Sigma-Aldrich) and vancomycin (Acros Organics) solutions, to which the microorganisms used were sensitive, were used as comparison drugs.
RESULTS AND DISCUSSION
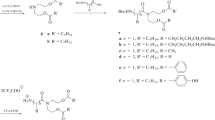
In this work, we proposed and implemented a scheme of the synthesis of novel cationic amphiphiles based on natural amino acids and diethanolamine (Scheme 1). The main criterion for the selection of synthesized amphiphile structures was the calculated value of hydrophilic–lipophilic balance (HLB). The HLB values correlate with the possibility of interactions of the therapeutic molecules with the components of the bacterial cell wall and the demonstration of antimicrobial activity. According to the published data for compounds exhibiting high antimicrobial activity, the optimal HLB ranges from 5 to 7 [17]. The theoretical calculation of the HLB was performed using the ACD/Labs, LogP software. A library of chemical structures based on amino acids and amino alcohols was developed and molecules whose HLB values lie within the range of probable antibacterial activity and range from 3.14 to 6.49 were selected (Table 1). To calculate the HLB in the chemical editor ChemDraw, we created the structures of the resulting compounds, which were then loaded into the LogP program for automatic calculation of the HLB according to the parameters set by the program. The calculation of values for molecules from literature sources [12, 17] with similar structure and with known antibacterial mechanism showed similar results. The data obtained became the basis for the development of the scheme for the preparation and synthesis of compounds 4(a–d) and 6(a–c).
All the compounds synthesized have a common principle of structure and consist of the following building blocks: two polar head groups, a spacer, a linker, and a hydrophobic alkyl fragment. The hydrophilic domain is represented by two residues of β-Ala, GABA, L-Phe, L-Tyr, or L-Trp. Diethanolamine was used as the spacer. Commercially available diethanolamine is often included in cosmetics and drugs because of its two reactive functional groups that allow the synthesis of preparations with a wide spectrum of action [18].

Scheme 1.
To form the hydrophobic domain of compounds 4(a–d) and 6(a–c), 1-bromoctane and 1‑bromododecane were proposed in this work. Lipophilic domain in 2(a, b) was obtained by the N-alkylation reaction of diethanolamine in the presence of K2CO3 in CH3CN. The yield of products 2a and 2b was 45% and 74%, respectively.
Compounds 3(a–d) containing Boc-protected aliphatic amino acids were prepared by the carbodiimide method using DCC and DMAP [19]. The yields of substances 3(a–d) were 40, 41, 62, and 60%, respectively. The structures of the obtained products were confirmed by 1H-NMR and IR spectroscopy.
Compounds 5(a–c) containing Boc-protected aromatic amino acids were prepared according to the method described above. The yields of compounds 5(a–c) were 72, 56, and 28%, respectively. The low yield of compound 5c containing tyrosine is explained by the fact that the presence of a reactive hydroxyl group leads to the formation of a complex mixture that is difficult to separate. The presence of many by-products makes it difficult to isolate the target compound with an unprotected OH group. A solution to this problem could be the use of commercially available derivatives with a protected hydroxyl group, such as tert-butyl protection.
Cationic amphiphiles 4(a–d) and 6(a–c) were obtained by removal of the Boc-protective groups by trifluoroacetic acid in anhydrous methylene chloride medium (1 : 1 v/v). The reaction was monitored by TLC data. The structures of the obtained salts were confirmed by mass spectrometry data.
The preliminary evaluation of antibacterial action of the synthesized compounds was performed on Gram-positive bacteria B. subtilis 534 and Gram-negative bacteria E. coli M17 by the method of bacteria diffusion in agar (Tables 2, 3). Compounds that contain the aliphatic amino acids β-Ala (4c) and GABA (4d) with a C12 hydrophobic chain in the polar group showed antibacterial activity against both Gram-positive and Gram-negative bacteria with a MIC of 1 μg/mL. Amphiphiles with the aromatic amino acids L-Phe (6a) and L-Trp (6b) in the polar head group and C8 hydrophobic chain were active against B. subtilis with a MIC of 1 µg/mL. Thus, the following structure-activity relationship can be noted: for high activity against Gram-positive bacteria B. subtillis, a pronounced hydrophobicity is required, which is achieved either by increasing the hydrocarbon chain length to C12 in the presence of aliphatic amino acids β-Ala and GABA, as in the case of compounds 4c and 4d, or by introducing hydrophobic aromatic acids L-Phe and L-Trp while maintaining the hydrocarbon radical C8, as in 6a and 6b. As for Gram-negative bacteria E. coli, this dependence is observed only for aliphatic amino acids with a long hydrophobic fragment, 4c and 4d, since the introduction of aromatic amino acids was not efficient with regard of the antibacterial activity.
REFERENCES
Yount N.Y., Yeaman M.R. 2004. Multidimensional signatures in antimicrobial peptides. Proc. Natl. Acad. Sci. USA. 101 (19), 7363. https://doi.org/10.1073/pnas.0401567101
Musin K.G. 2018. Antimicrobial peptides—a potential replacement for traditional antibiotics. Rus. J. Infection and Immunity. 8 (3), 295–308. https://doi.org/10.15789/2220-7619-2018-3-295-308
Rima M., Rima M., Fajloun Z., Sabatier J.-M., Bechinger B., Naas T. 2021. Antimicrobial peptides: A potent alternative to antibiotics. Antibiotics. 10 (9), 1095. https://doi.org/10.3390/antibiotics10091095
Molchanova N., Hansen P.R., Franzyk H. 2017. Advances in development of antimicrobial peptidomimetics as hotential drugs. Molecules. 22 (9), 1430. https://doi.org/10.3390/molecules22091430
Pirri G., Giuliani A., Nicoletto S.F., Pizzuto L., Rinaldi A.C. 2009. Lipopeptides as anti-infectives: A practical perspective. Cent. Eur. J. Biol. 4(3), 258–273. https://doi.org/10.2478/s11535-009-0031-3
Fjell C.D., Hiss J.A., Hancock R.E. W., Schneider G. 2012. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discovery. 11, 37–51.
Faber C., Stallmann H., Lyaruu D., Joosten U., Von Eiff C., van Nieuw Amerongen A., Wuisman P.I. 2005. Comparable efficacies of the antimicrobial peptide human lactoferrin 1-11 and gentamicin in a chronic methicillin-resistant Staphylococcus aureus osteomyelitis model. Antimicrob. Agents Chemother. 49 (6), 2438–2444. https://doi.org/10.1128/AAC.49.6.2438-2444.2005
Lin L., Chi J., Yan Y., Luo R., Feng X., Zheng Y., Xian D., Li X., Quan G., Liu D, Wu C., Lu C., Pan X. 2021. Membrane-disruptive peptides/peptidomimetics-based therapeutics: Promising systems to combat bacteria and cancer in the drug-resistant era. Acta Pharm. Sin B. 11 (9), 2609. https://doi.org/10.1016/j.apsb.2021.07.014
Tague A.J., Putsathit P., Hammer K.A., Wales S.M., Knight D.R., Riley T.V., Keller P.A., Pyne S.G. 2019. Cationic biaryl 1,2,3-triazolyl peptidomimetic amphiphiles: Synthesis, antibacterial evaluation and preliminary mechanism of action studies. Eur. J. Med. Chem. 168, 386. https://doi.org/10.1016/j.ejmech.2019.02.013
Mojsoska B., Jenssen H. 2015. Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals (Basel). 8(3), 366–415. https://doi.org/10.3390/ph8030366
Zhang E., Bai P.-Y., Cui D.-Y., Chu W.-C., Hua Y.-G., Liu Q., Yin H.-Y., Zhang Y.-J., Qin S., Liu H.-M. 2018. Synthesis and bioactivities study of new antibacterial peptide mimics: The dialkyl cationic amphiphiles. Europ. J. Med. Chem. 143, 1489–1509. https://doi.org/10.1016/j.ejmech.2017.10.044
Su M., Xia D., Teng P., Nimmagadda A., Zhang C., Odom T., Cao A., Hu Y., Cai J. 2017. Membrane-active hydantoin derivatives as antibiotic agents. J. Med. Chem. 60 (20), 8456. https://doi.org/10.1021/acs.jmedchem.7b00847
Konai M.M., Ghosh C., Yarlagadda V. 2014. Membrane active phenylalanine conjugated lipophilic norspermidine derivatives with selective antibacterial activity. J. Med. Chem. 57, 9409–9423. https://doi.org/10.1021/jm5013566
Ghosh C., Sarkar P., Samaddar S., Uppua D., Haldar J. 2017. L-Lysine based lipidated biphenyls as agents with anti-biofilm and anti-inflammatory properties that also inhibit intracellular bacteria. Chem. Commun. 53, 8427–8430. https://doi.org/10.1039/C7CC04206J
Lohan S., Kalanta A., Sonkusre P., Cameotra S. S., Bisht G. S. 2014. Development of novel membrane active lipidated peptidomimetics active against drug resistant clinical isolates. Bioorg. & Med. Chem. 22, 4544–4552. https://doi.org/10.1016/j.bmc.2014.07.041
Schnaider L., Brahmachari S., Schmidt N.W., Mensa B., Shaham-Niv S., Bychenko D., Adler-Abramovich L., Shimon L.J.W., Kolusheva S., DeGrado W.F., Gazit E. 2017. Self-assembling dipeptide antibacterial nanostructures with membrane disrupting activity. Nat. Commun. 8 (1), 1365. https://doi.org/10.1038/s41467-017-01447-x
Shahane G., Ding W., Palaiokostas M., Azevedo H.S., Orsi M. 2019. Interaction of antimicrobial lipopeptides with bacterial lipid bilayers. J. Membr. Biol. 252 (4–5). 317. https://doi.org/10.1007/s00232-019-00068-3
Yar M., Mushtaq N., Afzal S. 2013. Synthesis, reactions, applications, and biological activity of diethanolamine and its derivatives. Russ. J. Org. Chem. 49 (7) 949–967. https://doi.org/10.1134/S1070428013070014
Denieva Z.G., Romanova N.A., Bodrova T.G., Budanova U.A., Sebyakin Yu.L. 2019. Synthesis of amphiphilic peptidomimetics based on the aliphatic derivatives of natural amino acids. Moscow Univ. Chem. Bull. 74 (6), 300–305. https://doi.org/10.3103/S0027131419060087
Makovitzki A., Baram J., Shai Y. 2008. Antimicrobial lipopolypeptides composed of palmitoyl Di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry. 47 (40), 10630. https://doi.org/10.1021/bi8011675
Funding
The work was supported by the Russian Foundation for Basic Research (project no. 20-04-00672). The work was performed using the equipment of the Center for Collective Use of MIREA – Russian technology university supported by the RF Ministry of Education and Science (Agreement no. 075-15-2021-689 dated September 1, 2021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A. Dunina-Barkovskaya
Rights and permissions
About this article

Cite this article
Guseva, M.K., Denieva, Z.G., Budanova, U.A. et al. Cationic Lipoaminoacid Derivatives of Diethanolamine As Potentially Membrane-Active Antibacterial Agents. Biochem. Moscow Suppl. Ser. A 17, 148–155 (2023). https://doi.org/10.1134/S1990747823020034
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990747823020034