
Abstract—
Glycoproteins IIb-IIIa (GPIIb-IIIa), also known as αIIbβ3 integrins, are key platelet adhesion receptors. These molecules are the most abundant (over 10 000 copies per cell) transmembrane receptors playing a crucial role in thrombus formation by promoting platelet aggregation. Integrins need to undergo activation and transit to high-affinity state for their ligands – fibrinogen, fibrin, and von Willebrand factor (VWF) – in order to form bonds between platelets. Activation of integrins is mediated by a set of various messengers through intracellular signalization. Integrins αIIbβ3, like other integrins, are capable of reverse signal transmission inside the cell, called “outside-in” signaling. Recent studies have shown heterogeneity of the thrombus structure and the existence of a stable and dense inner core and a fluid-like loose shell. Since platelet aggregation is provided by integrin-mediated interactions, one can suggest that it is the features of integrin activation and clustering that strongly influence the formation of thrombus architecture. This work is intent on systematizing recent data concerning activation and functioning of platelet integrins αIIbβ3 and searching for correlations between thrombus heterogeneity and the state of integrins on the platelets surface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 INTRODUCTION
Integrins are cell-surface transmembrane receptors responsible for intercellular binding [1]. Cell aggregation and adhesion to the vessel wall is mediated by this membrane protein family [2]. Integrins are heterodimeric proteins that consist of noncovalently associated α and β subunits. Upon activation α- and β-chains change interposition that results in an enhanced ligand affinity. Thus, integrins are able to exist in low-affinity and high-affinity states. Intermediate activation state has also been reported [3, 4]. Like the other adhesive receptors, integrins are closely associated with the cytoskeleton and thus are capable of mediating whole cell adhesion. However, the ability to change the ligand affinity as well as signal transduction through the cell membrane both ways is a unique feature of integrin family proteins.
Integrins provide aggregation of platelets–blood cells responsible for the integrity of the circulatory system. This class of adhesive receptors plays a pivotal role in the hemostasis (the process that causes hemorrhage to stop) and thrombosis [2, 5, 6]. Upon vessel wall damage and vascular endothelium dysfunction, platelets become activated as a result of interaction with extracellular matrix proteins (collagen, fibronectin, laminin) and/or soluble agonists (ADP, thrombin, adrenaline, thromboxane) [7]. Activated platelets adhere to the breach in the vessel wall, form a hemostatic plug and thus abrogate blood loss. Whole variety of integrins is present on the platelets surface: α2β1, α5β1, α6β1 (β1 subfamily), αLβ2 (β2 subfamily), αIIbβ3, ανβ3 (β3 subfamily). Only one of the major platelet integrins, αIIbβ3 (GPIIb-IIIa), is able to switch to the high-affinity state during activation. Furthermore, αIIbβ3 is present in the highest number of copies–100 000 per cell [8–10].
In an activated state αIIbβ3 is capable of binding fibrinogen and von Willebrand factor (vWF) molecules, which are both present in in blood plasma and can be released from the platelet alpha granules [11] during platelet activation. vWF can be released by the activated endothelial cells [12–14]. Other ligands – vitronectin, fibronectin, and thrombospondin regulate platelet adhesion to the subendotheial matrix exposed in case of vessel wall damage [14]. Activated platelets aggregate via αIIbβ3-fibrinogen/vWF-αIIbβ3 bonds forming a thrombus which can then cover the breach or occlude the whole vessel [6, 15].
Arterial thrombus formed in microvessel has a heterogeneous structure. Highly activated platelets are located close to the injury. They release granules content (soluble activators), which provides further clot growth. These platelets are closely packed, forming a stable thrombus core [16, 17]. Platelets of the outer part are less activated and weakly connected, forming the thrombus shell. The shell is loosely packed and can “flow” around the dense core. One can assume that there is a significant difference in adhesion forces, which results in the gradient of packing density. In its turn, the extent of interplatelet interaction can be varied by the strength of single bonds or local increases of bonds density. αIIbβ3 is the most potent candidate for being a regulator of this process.
Platelet activation causes integrin ligand affinity change via an “inside-out” signaling pathway. It has also been proposed that the integrin activation can be induced by the ligand itself [18]. Inside-out activation requires changes in the subcellular localization of Talin-1, Kindlin-3 and the small GTPase Rap1b [19–22]. Rap1b activation implies the exchange of the bound GDP to GTP. This process engages two important secondary messengers of the platelet signaling – Ca2+ and phosphatidylinositol (3,4,5)-trisphosphate. αIIbβ3 cytoplasmic tails can be associated with actin-bound Talin-1 and Kindlin. Thereby integrin activation can be induced by the lateral stresses of membrane resulting in the impact on the actin cytoskeleton [23]. αIIbβ3 interaction with its ligands leads to the further enhancement of platelet activation (“outside-in” activation). Outside-in activation can be experimentally observed in the process of platelet spreading on fibrinogen-coated surface [18, 24]. Integrin-dependent signaling is known to depend on Syk tyrosine kinase usually activated by ITAM-bearing receptors [25]. αIIbβ3 has no ITAM domain, therefore it is still under discussion which protein acts as a mediator and if this mediator is required [26–29].
Integrin αIIbβ3 is also found in megakaryocytes, mast cells, basophils and some tumor cells (melanomas, squamous cell carcinomas, etc.) [30–36]. Nevertheless, binding to anti-CD61 and anti-CD41 (antibodies against β3 and αIIb chains) are often used as platelet markers. Outside-in or inside-out signaling disruptions as well as platelet αIIbβ3 deficiency (Glanzmann thrombasthenia) lead to severe hemostatic disorders: menorrhagia, nosebleeds, gastrointestinal bleedings, postpartum hemorrhages, and postoperative bleeding [37, 38]. In patients with Glanzmann thrombasthenia thrombus growth on collagen is disturbed at any shear rates, while in patients with Bernard–Soulier syndrome (vWF receptor GPIb deficiency) clot formation is normal at low shear rates [39].
The aim of this review is a survey of the knowledge on platelet αIIbβ3 functioning, the influence of weak and strong platelet activation at αIIbβ3 clustering, and the role of integrins in the process of the thrombus heterogeneous structure formation.
2 MOLECULAR EVENTS OF αIIbβ3 ACTIVATION
2.1 Structure of Integrin αIIbβ3
Integrins are integral plasma membrane glycoproteins composed of α and β chains. Each subunit consists of a large extracellular domain, a transmembrane region, and a short cytoplasmic sequence of amino acids. Extracellular domain of the αIIb chain consists of β-propeller–the structure of a series of repeats, which forms a seven-pointed (seven blades) circularly threaded sequence with a central cavity [40, 41]. Aspartate bonded cations increase the rigidity of the interface between β-propeller and underlying domains (thigh, calf1, calf2). Flexible hydrophobic “genu” between the thigh and the calf1 allows to change the interposition of the αIIb and β3 chains [42–44] (Fig. 1a).

Platelet αIIbβ3–integrin structure. (a) Extracellular domain of the clasped αIIbβ3 integrins (PDB : 3FCS). Three metal binding sites are depicted in domain I of the β3-chain. (b) Aminoacid residues adjoining the metal-binding sites. (c) Unclasped form of the αIIbβ3 integrin bound to ligand (PDB : 3FCS).
Chain β3 consists of the A-domain, which extends into the cavity of the αIIb subunit propeller by virtue of arginine (Arg261) [42, 45–47]. A-domain has several metal binding domains (MIDAS, ADMIDAS, LIMB, see Fig. 1b) [48] and also binds EGF-domains via hybrid domain. EGF domains are rich in cysteines and form rod-shaped structure via disulfide bonds [49, 50]. A flexible hydrophobic “genu” is also present between EGF and hybrid domains as in the αIIb chain [46, 51].
Transmembrane domain of αIIbβ3 consists of two crossed helixes. αIIb also contains GFFKR sequence in its transmembrane domain. In inactivated αIIbβ3 this sequence is hidden in the plasma membrane (Fig, 1a), but when the receptor conformation changes, GFFKR goes to the cytosol and becomes a docking site for multiple effector proteins [53, 54] (Fig. 1b).
Cytoplasmic domains of both α and β subunits are small sequences, which include functional motifs, such as NxxY [55, 56].
Integrins αIIbβ3 are able to bind a number of ligands: fibrinogen, fibrin, VWF. They bind different sequences of fibrinogen (KQAGDV) and VWF (RGD). MIDAS and ADMIDAS cation binding domains play an important role in ligand coordination [57].
Due to flexible “genu” domains of αIIb and β3 subunits, integrins can change conformation and switch from the closed (low-affinity) to the open (high-affinity) state.
2.2 Membrane-Dependent Events of Integrin Activation
Cytoplasmic domain of the β3 subunit contains two NxxY motifs, which serve as the docking sites for Talin-H and kindlin-3 which are known to lead to the conformational changes in αIIbβ3. Inactive Talin-H is associated with Talin-R and cannot bind β3 subunit. Couple of Talin subunits can be separated by a set of different pathways. High Ca2+ concentration activates calpain, which cleaves the bond between Talin-H and Talin-R and makes Talin-H active. On the other hand, Talin subunits can be separated as a result of Talin and RIAM interaction [58, 59].
FERM domains of the free Talin-H bind NxxY domains on the αIIbβ3. This is proposed to be one of the main stages of αIIbβ3 activation. Talin-H can link integrins with an actin cytoskeleton directly or via vinculin, thus promoting platelet shape change. Talin-H is able to interact with membrane phosphoinositides (e.g., PIP2), inactivating in the process [60]. The importance of Talin was demonstrated in the studies of integrin- and Talin-contrasfected CHO cells: in the absence of Talin integrins did not switch to the active state [55, 61].
The kindlin family of proteins also plays a significant role in αIIbβ3 activation, since neither the inside-out nor the outside-in signaling works without them [62, 63]. Like talin, kindlin interacts with NxxY motif of β3 integrin subunit (often with the one which is distant from the membrane) via FERM-domain. Kindlin also holds PH-domain, which associates with membrane phosphoinositides [62, 63]. Nonetheless, the exact mechanism of αIIbβ3 activation mediated by kindling-3 remains unclear [64].
Filamin and ICAP proteins can block integrins activation by competing with talin and kindlin [65]. Migfilin displaces filamin from the cytoplasmic domain of the β3 subunit serving as a cofactor of integrin activation, in particular kindling-2 cofactor, as it was demonstrated in CHO cells.
CIB [66], ICln [67], Aup1 [68], PP1 phosphatase [69], and sharpin [70] are able to bind αIIb, but their roles remain unspecified.
2.3 Integrin Inside-Out Signaling
2.3.1. GPCR-induced activation. The majority of the platelet receptors, including thrombin-activated PAR1 and PAR4, as well as ADP-activated P2Y1 and P2Y12, transmit cellular signals through G-proteins (GPCRs, G-protein-coupled receptors) [71]. GPCR binding to its ligand leads to the GDP-to-GTP exchange in the Gα subunit of heterotrimeric G-protein and complex dissociation, resulting in the appearance of Gβɣ and Gα-GTP signal transmitters. Gβγ and Gα-GTP of class q can activate phospholipase Cβ (PLCβ) together or independently [72]. Moreover, Gα-GTP of class i and z, produced upon platelet activation, inhibit adenylate cyclase, thus lowering cAMP concentration and therefore enhancing the PLCβ activation [73]. Gβγ, which is present in 50 000 copies per platelet [74], is capable of activating PI3Kγ.
2.3.2. Tyrosine-kinase induced activation. Upon adhesion to a damaged wall of a vessel, platelets interact with the components of extracellular matrix protein collagen. Collagen activates platelet GPVI receptors. This induces a tyrosine-kinase signaling cascade, based on Syk and SFK kinases [76–78]. After the GPVI activation by collagen, SFK phosphorylates two tyrosine-containing YxxL motifs in the Fcγ-chain (ITAM), which is covalently bound to GPVI. Syk binds phosphorylated YxxL motifs by its SH-2 domains that turn into an active state. Active Syk-kinases phosphorylate large adaptor protein LAT, which rapidly forms complexes with PI3Kβ and PLCγ2 [79]. PI3Kβ and PI3Kγ produce PIP3 from PIP2. PIP3 serves as a docking site for a set of PH-domain-containing proteins, among which are Btk and the above mentioned kindlin. Activated PLCγ2 and PLCβ hydrolize PIP3 and produce IP3 and DAG [25, 80, 81]. Signaling via other tyrosine-kinase-dependent receptors CLEC-2 and FcγRIIa, which act in a more specific physiological settings, closely resembles GPVI.
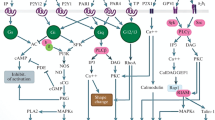
2.3.3. Rap1, RASA3, CalDAG-GEFI. IP3 and DAG produced by PLC are important secondary messengers of the platelet activation. IP3 binds IP3 receptors on the surface of the dense tubular system (DTS, a remnant of the megakaryocyte endoplasmic reticulum). This leads to a release of free calcium ions into the platelet cytosol [71, 84]. DAG activates PKC–a serine kinase capable of phosphorylating multiple targets in a cell. An increase in the concentration of free calcium ions results in an activation of not only calpain, but also of CalDAG-GEFI [85]. This protein catalyzes reaction of the exchange of GDP on GTP in the nucleotide binding site of Rap1b. Active Rap1b together with RIAM and talin-H leads to the activation of αIIbβ3 integrin [22, 58, 86]. Rap1b-GTP can inactivate spontaneously as a result of GTP hydrolysis. This reaction can be significantly enhanced upon binding of Rap1b by GAP-proteins (GTPase activating proteins). Major platelet GAP is RASA3 [87, 88]. RASA3 contains PH-domain that can bind with PIP2 and PIP3. This changes cellular localization of RASA3. Thus, after PIP3 production, RASA3 leaves integrin-enriched areas, which increases Rap1b-GTP lifetime and results in the αIIbβ3-integrin activation [87, 88]. Furthermore, it is hypothesized that phosphorylation of CalDAG-GEFI by PKA (which is inhibited upon platelet activation) prevents the CalDAG-GEFI-mediated GDP–GTP exchange in the Rap1b nucleotide binding site. Thus, integrin activation pathway is inhibited in resting platelets (see Fig. 2a) [59, 85].

Schematic representation of platelet intracellular signaling, mediating and mediated by αIIbβ3 integrins. (a) “Inside-out” αIIbβ3 integrin activation; (b) platelet signaling amplification scheme (“outside-in” signaling).
2.3.4. Role of platelet interaction with vWF in the integrin activation. Vessel wall damage in not limited to the extracellular matrix proteins exposure. Another major outcome of this process is an activation of the endothelial cells, which release Weibel–Palade bodies containing vWF. vWF is a multimeric protein, which can be formed by dozens of 250-kDA monomers [89, 90]. vWF was also reported to be present in the blood plasma and platelet α-granules [91]. High shear rates in the blood flow (alongside with the turbulent flows) result in the unfolding of vWF multimers bound to collagen or endothelial cells. vWF unfolding exposes its A1 domains that bind platelet GPIb receptors. This is the main event in the initial platelet adhesion and, probably, further platelet activation via tyrosine-kinase signaling pathway (Syk and SFK) [92]. vWF also binds active αIIbβ3-integrins by its C1 domains. This induces integrin outside-in signaling (see Section 2.4), amplification of the platelet activation, and further platelet aggregate formation at sites of vessel wall breach [93].
2.4 Activation Amplification: Outside-In Signaling
After activation and transition into a high-affinity state, platelet integrins αIIbβ3 can bind fibrin, fibrinogen, or vWF. This results in an additional amplification of platelet activation–outside-in signaling [94]. As was mentioned above, cytoplasmic domains of activated integrins are bound to actin cytoskeleton via talin and kindlin complexes. Upon cell activation, most of the signaling molecules relocate by means of diffusion or actin cytoskeleton reconstruction. Thus integrins can form heteroclusters with other receptors, such as GPIb, GPVI, FcγRIIa [24, 27, 28, 95]. SFK kinases are bound to polyproline amino-acid regions in the cytoplasmic regions of these receptors via SH-3 domains [96, 97]. Thus, active SFK are located in the close proximity of activated αIIbβ3 integrins. SFK phosphorylates tyrosine residues in the integrin NxxY motifs, which results in the additional activation of Syk and SFK kinases. This amplifies the platelet activation. It was demonstrated many times that GPVI or FcγRIIa deficiency results in disordered activation of integrin αIIbβ3 and disturbance in the thrombus growth in mice. On the other hand, clustered αIIbβ3 integrins can activate Syk-kinases in the absence of GPVI or FcγRIIa [27, 28]. Furthermore, tyrosine residues phosphorylation in the cytoplasmic domain of the β3‑chain is not required for Syk activation. The prime hypothesis is that Syk can associate with the cytoplasmic domain of the β3-chain of αIIbβ3 integrin and activate via autophosphorylation or trans autophosphorylation [29]. Positive feedback loop, produced by the outside-in signaling significantly amplifies platelet activation (see Fig. 2b).
2.5 Platelet Integrin Clustering and Irreversible Fibrinogen Binding
As was mentioned previously, platelet integrin clustering is required for an effective platelet activation amplification (outside-in signaling). However, platelet integrin clustering role in the process of thrombus growth remains unclear. Integrin clustering can be observed by binding of exogenous human plasma fibrinogen, conjugated with fluorescent or radioactive label [98]. For experimental assessment, washed platelets from suspension or platelets from the whole blood are immobilized on a glass, covered by an adhesive protein [89, 99].
Pictures of binding of FITC-conjugated exogenous human serum fibrinogen by washed platelets are given in Fig. 3 [100]. In the first experiment (Figs. 3a and 3b) platelets were spread on fibrinogen. Irreversible fibrinogen binding with integrin clusters can be observed (fibrinogen clusters remain bound to platelets after washing by the solution, Fig. 3b), while fibrinogen binding to single integrin molecules is disrupted upon cell suspension dilution (fibrinogen clusters in Fig. 3b are more contrast than in Fig. 3a). In the experiment illustrated in Figs. 3c and 3d, platelets were spread on the anti-PECAM1-receptor antibodies [101]. Platelets were then activated by 10 µM of ADP. It can be seen that platelet spreading correlates with the integrin cluster formation (in Fig. 3c a spread platelet binds fibrinogen, while a non-spread platelet in Fig. 3d, does not).

Fluorescently labeled fibrinogen binding by human blood platelets: microscopy. Upper image, in differential interference contrast, lower image, FITC fluorescence (epifluorescence). (a, b) Washed platelets were perfused by syringe pump through the flow chamber with fibrinogen coated glass (100 µg/mL). Non-immobilized platelets were washed by Tyrode’s buffer for 20 min. This was followed by application of 10 µM ADP solution and FITC-labeled fibrinogen (100 µg/mL) [100]. Fibrinogen binding was observed by epifluorescence. This was followed by the perfusion with Tyrode’s buffer and washing away non-bound fibrinogen. (a) Platelet before washing off of fibrinogen; (b) platelet after fibrinogen was washed off. Irreversible fibrinogen binding was observed in a form of high intensity fluorescence “clusters”. (c, d) Washed platelets were perfused by syringe pump through the flow chamber with anti-PECAM1 antibodies (VM64, 20 µg/mL). Non-immobilized platelets were washed by Tyrode’s buffer for 20 min. This was followed by perfusion with 10 µM ADP solution and FITC-labelled fibrinogen (100 µg/mL) [100]. Activated (spread) platelets bound fibrinogen (c), while non-activated platelets (with lamellipodiae), did not (d).
Figure 4 presents flow cytometry data. In the experiment illustrated in Fig. 4a, platelet fibrinogen binding upon activation by thrombin and TRAP-6, followed by a twofold dilution by the buffer, was assessed. Dilution of the cell suspension resulted in a decrease of fibrinogen binding upon weak agonist stimulation (TRAP-6). Thus, at specific conditions, fibrinogen binding by platelet integrins (or at least by some integrin domain) is reversible. Upon stimulation of platelets (2 × 107 cells/mL) by ADP and a further 20-fold dilution of the suspension with a fibrinogen-free buffer, a clear separation of platelets into two subpopulations differing in binding of labeled fibrinogen was observed (Fig. 4b). It is likely that one of the populations (with low integrin activation degree) binds fibrinogen reversibly, while the other one, irreversibly [100].

Flow cytometry of FITC-labelled fibrinogen binding to platelets in a diluted cell suspension. Platelets were washed as was described previously [71]. (a) Continuous flow cytometry performed as described elsewhere [71, 128]: time dependence of FITC fluorescence intensity in arbitrary units. Platelet concentration was 106 cells/mL. Fibrinogen was added before the experiment (data not shown). After that, activators (thrombin, 25 nM, black curve; TRAP-6, 10 µM, gray curve) were added to the cell suspension. After a twofold dilution by fibrinogen-free solution, a decrease in fibrinogen binding is observed for weak activation (TRAP-6). (b) Fibrinogen binding by platelets in cell suspension (2 × 107 cells/mL) before and after the ADP-induced stimulation (10 µM).
These results may suggest that there is a heterogeneity in platelet activation state, which corresponds to the ligand–integrin activation degree: single integrin molecules that bind fibrinogen reversibly and integrin clusters that bind fibrinogen irreversibly. It is noteworthy that in a recent report of researchers of the Technological University of Georgia at ISTH SSC 2018 meeting, the existence of a new “not complete activation” state of platelets was proposed independently; this state can be induced by mechanoreception via glycoprotein Ib. It is proposed that this state of platelet activation corresponds to a low-affinity binding of integrins to fibrinogen and fibronectin [102]. However, this suggestion requires additional confirmation, since it contradicts previously published data [103–106] on binding of fibrinogen of activated integrin antibodies (PAC-1) by activated platelets in cell suspension. Only one type of fibrinogen binding sites was described. It should also be taken into account that receptors bound with fibrinogen are mostly clustered [107].
3 MECHANICS OF THE αIIbβ3–LIGAND INTERACTIONS
Platelet–platelet interactions should be strong enough to resist flow forces acting on a growing thrombus. Atomic force microscopy shows that the force between platelets can reach a value of nanonewtons [108].
The mechanics of binding to its ligands was studied by the group of R. Litvinov et al. [94, 109, 110] by means of an optical tweezers – a highly focused laser beam, which allows manipulating small dielectric objects. Optical tweezers can be also used for measuring the force applied to the held object, which is proportional to the magnitude of its displacement from the tweezer center. For the experiments, they utilized silica pedestals coated with various αIIbβ3 ligands. Silica pedestal was brought into contact with latex bead coated with purified human αIIbβ3 receptors [94, 110].
The study on interaction of integrins with fibrinogen revealed that the critical force distribution has two modes, being a sum of exponentially decreasing (up to 20–50 pN) and normally distributed (peaked at 70–80 pN) parts (Fig. 5a). The bimodal nature of the distribution possibly suggests that αIIbβ3–fibrinogen complex exists in two bound states with different stability [94].

Monomeric fibrin (m-fibrin) displayed a significantly higher probability of interaction with αIIbβ3 than fibrinogen (Fig. 5b). However, the disruption force peaks were barely distinguishable and the mean force was much higher, thus one can suggest that the interactions could be multimolecular. The comparison between the critical forces for m-fibrin and fibrinogen should be qualitative. It also should be taken into account that the inhibition by 1 mM of γC-dodecapeptide (H12) abolished the second mode of αIIbβ3–fibrinogen interaction. With the 1 mM of cRGD added for both fibrin and fibrinogen only exponentially decreasing part of force distribution was observed. This may indicate that the critical force and the number of possible states varies for integrins interacting with different ligands [94]. For interaction of αIIbβ3 with polymerized fibrin, one can distinguish two peaks in critical force distribution, with mean value higher than that for other ligands [110].
Thus, the critical forces for the αIIbβ3–ligand complexes should be ranged in the following order: fibrin polymer > fibrin monomer > fibrinogen (Fig. 5b). Integrin–ligand complex presumably exists in two states with different stability. Nevertheless, we cannot specify, to what extent the forces between platelets change during the activation, as the isolated receptors were studied, so it is not possible to analyze their state.
Earlier Litvinov et al [111] investigated platelet interaction with fibrinogen. The distribution of the critical forces for ADP-activated platelets was also found to be bimodal, but the first peak was interpreted as a non-specific interaction, since it was detected for unstimulated platelets. At the same time, we can suggest that some integrins of these platelets were also activated, as SEM image showed that the platelets were balloon-shaped and had filopodia [111]. Therefore, this result cannot be interpreted unambiguously. On the other hand, earlier studies [103–106] investigated platelet interaction with fibrinogen and PAC-1 (the antibody to activated αIIbβ3) in suspension and reported the existence of a single binding site type.
4 PLAUSIBLE ROLE OF αIIbβ3 IN THE THROMBUS STRUCTURE
The development of the murine models of the in vivo thrombus formation has taken studies of the thrombus structure and dynamics to the next level. The use of fluorescent labeling and confocal microscopy makes it possible to investigate the details of the thrombus formation process in the damaged thin-walled microvessel with a high resolution.
This is how it was shown that the microvascular thrombus has a heterogeneous structure (Fig. 6): a stable part is adjacent to the injury, while outer layers are fluid-like [7]. The analysis of intrathrombus transport of fluorescently labeled albumin revealed that the inner part of the thrombus is more dense (less albumin is observed in the gaps between cells). One can suppose that platelet packing density heterogeneity is caused by the heterogeneity of the forces. Indeed, platelets and their adhesive receptors differ in the extent of activation in different parts of the clot: P-selectin positive degranulated platelets are localized close to the damaged vessel wall [7, 16] there fibrin is also found. Dense, stable and highly activated part is called the core; loose, fluid-like part of slightly activated platelets is called the shell [7].

Model of a microvascular thrombus. Microvascular thrombus has a heterogenous structure: it consists of a dense core that contains highly activated platelets and a fluid shell composed of weakly activated cells. Core platelets release ADP from their dense granules. Density of the core accounts for a impeded diffusion of ADP and thrombus from the core to the shell (thrombin gradient is depicted by a darker color). Fibrin is localized at the thrombus base. It is assumed that αIIbβ3-integrins in the platelet core are clustered. This amplifies platelet activation (granule release, transition to a procoagulant state) and strengthening of the platelet–platelet bonds. Core platelets also change shape (not shown in the figure for simplification).
It is believed that the core requires thrombin generation – in the presence of its inhibitor the core is abolished, the thrombus itself is unstable and undergoes cycles of growths and disruptions [7, 16]. It is also assumed that the packing density heterogeneity leads to the gradient of thrombin concentration. Thrombin is retained in the core – this, on the one hand, limits the growth of a clot, but on the other, contributes to the polymerization of fibrin in an already formed core [7, 17].
Platelet activation via ADP receptor P2Y12 is critical for the formation of thrombus shell. In the experiments in the presence of cangrelor (P2Y12 inhibitor) the size of the shell was significantly reduced while the core remained unaffected. At the same time, for mice with Gi2 gain of function mutation, the shell but not the core was reduced in size. This mutation eliminated negative feedback during P2Y12-induced signaling (usually provided by the RGS proteins-dependent inactivation). Thus, it is shown that the P2Y12 signaling is important for the formation of the thrombus shell [7]. As mentioned above, P2Y12 is associated with Gi-protein; P2Y12 activation leads to protein kinase A inactivation and PI3Kγ activation, that is additional inside-out integrin activation. It is likely that thromboxane A2, the other secondary mediator if secondary activation, has a significant role in the formation of thrombus shell.
One can assume that ADP concentration on the outer thrombus layers is sufficient only for reversible or partly reversible activation of a limited number of platelet integrins (weak activation, similarly to Fig. 4a, TRAP-6). It may be due to the fact that the loose thrombus shell allows soluble agonists leakage or that the required ADP concentration cannot be provided by its sources (e. g., degranulated platelets). That is why the shell is unstable. Thrombin leads to strong platelet activation and fibrin formation near the injury – fibrin itself binds αIIbβ3 and strengthens the thrombus. At the same time thrombin distribution is restrained, limiting thrombus growth.
5 INACTIVATION OF PROCOAGULANT PLATELET INTEGRINS
Upon strong activation [112], platelets can turn into procoagulant ones, undergoing necrosis-like [113, 144] programmed cell death [84]. Negatively charged phosphatidylserine (PS) externalizes in the outer membrane of these platelets, enhancing the assembly of procoagulant complexes [115, 116]; other specific reactions also take place [117, 118]. Interestingly, integrins are inactive in procoagulant platelet subpopulation [119]. Presumably, during this process there occurs integrins activation and subsequent inactivation, but the mechanism is still unclear [120]. It is assumed that calpain cleavage of integrin-associated cytoskeletal proteins and phospholipid scrambling both contribute to integrin inactivation [121], although in some works calpain inhibition did not affected this phenomenon [122]. Remarkably, procoagulant platelets remain attached to the thrombus [123, 124]. This interaction is provided [125] by alpha-granule protein-covered “cap” formation mediated by transglutaminase activity and fibrin polimerization [126]. The cap promotes binding to adjacent platelets.
6 CONCLUSIONS
Integrins αIIbβ3 are platelet receptors that mediate stable aggregation. They can change conformations – presumably there is a spectrum of states with different affinity to ligands (fibrinogen, fibrin, von Willebrand factor). For integrin to switch to high-affinity state Ca2+- and phosphoinositide-dependent activation of Rap1b GTPase is required. Some studies [43, 53] consider stress-dependent integrin-associated actin cytoskeleton rearrangement as alternative pathway of the αIIbβ3 activation. Weak platelet activation results in reversible integrins binding (similarly to Fig. 4a, TRAP-6), while strong activation leads to irreversible. The integrins clustering seems necessary and sufficient for the occurrence of outside-in signaling and platelet activation amplification.
The ability of integrins to exist in different states (activated and inactive) is of great importance for the formation of thrombus heterogeneous structure. For the core, thrombin is critical, just as ADP is important for the shell. It can be assumed that integrins of thrombus shell platelets weakly interact via fibrinogen because ADP cannot induce intensive enough αIIbβ3 activation and clustering. On the other hand, a large fraction of integrins on the activated platelets of the thrombus core is clustered and αIIbβ3–fibrinogen/fibrin binding is irreversible. The complex of integrin–ligand clusters is stable and can resist high forces probably due to the increase of local receptor density. Therefore, the stability and high density of the inner thrombus part is ensured. Inflated packing density of the core prevents thrombin leakage, on the one hand, promoting its high local concentration and fibrin polymerization and on the other hand, confines thrombus core growth. Strongly activated platelets release ADP from dense granules, providing weak activation of platelets of the clot outer layers (the concept is shown in Fig. 6) [7, 16, 17].
Further studies require the development of new methods which would allow the measurement of critical platelet integrin–ligand forces. Nowadays the data for isolated receptors is available, thus only the order of magnitude can be estimated. It is impossible to compare values for integrins of different activation extent or evaluate clustering contribution.
It was shown that fibrin binds αIIbβ3 stronger than fibrinogen [110]; it is necessary to analyze its input into the stability of the thrombus in vivo. This would allow making a conclusion on whether fibrin can be used as a pharmacological target for clot destabilization.
ACKNOWLEDGMENTS
This work is supported by the Russian Science Foundation (project no. 17-74-20045).
7 COMPLIANCE WITH ETHICAL STANDARDS
Conflict of interests. The authors declare that they have no conflict of interest.
Statement of compliance with standards of research involving humans as subjects. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants involved in the study.
REFERENCES
Bouvard D., Brakebusch C., Gustafsson E., Aszódi A., Bengtsson T., Berna A., Fässler R. 2001. Functional consequences of integrin gene mutations in mice. Circ. Res. 89 (3), 211–223.
Yakimenko A.O., Sveshnikova A.N., Artemenko E.O., Panteleev M.A. 2014. This mysterious platelet. Priroda (Rus.). (2002), 3–8.
Bennett J.S. 1990. The molecular biology of platelet membrane proteins. Semin. Hematol. 27 (2), 186–204.
Plow E.F., Byzova T. 1999. The biology of glycoprotein IIb-IIIa. Coron. Artery Dis. 10 (8), 547–551.
Stalker T.J., Newman D.K., Ma P., Wannemacher K.M., Brass L.F. 2012. Platelet signaling. Handb. Exp. Pharmacol. (210), 59–85.
Panteleev M.A., Ananyeva N.M., Greco N.J., Ataullakhanov F.I., Saenko E.L. 2005. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J. Thromb. Haemost. 3 (11), 2545–2553.
Stalker T.J., Traxler E.A., Wu J., Wannemacher K.M., Cermignano S.L., Voronov R., Diamond S.L., Brass L.F. 2013. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 121 (10), 1875 –1885.
Niiya K., Hodson E., Bader R., Byers-Ward V., Koziol J.A., Plow E.F., Ruggeri Z.M. 1987. Increased surface expression of the membrane glycoprotein IIb/IIIa complex induced by platelet activation. Relationship to the binding of fibrinogen and platelet aggregation. Blood. 70 (2), 475–483.
Wagner C.L., Mascelli M.A., Neblock D.S., Weisman H.F., Coller B.S., Jordan R.E. 1996. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood. 88 (3), 907–914.
Stouffer G.A., Smyth S.S. 2003. Effects of thrombin on interactions between beta3-integrins and extracellular matrix in platelets and vascular cells. Arterioscler. Thromb. Vasc. Biol. 23 (11), 1971–1978.
Harrison P., Cramer E.M. 1993. Platelet alpha-granules. Blood Rev. 7 (1), 52–62.
Plow E.F., Haas T.A., Zhang L., Loftus J., Smith J.W. 2000. Ligand binding to integrins. J. Biol. Chem. 275 (29), 21785–21788.
Plow E.F., D’Souza S.E., Ginsberg M.H. 1992. Ligand binding to GPIIb-IIIa: A status report. Semin. Thromb. Hemost. 18 (3), 324–332.
Savage B., Cattaneo M., Ruggeri Z.M. 2001. Mechanisms of platelet aggregation. Curr. Opin. Hematol., 8 (5), 270–276.
Panteleev M.A., Ananyeva N.M., Ataullakhanov F.I., Saenko E.L. 2007. Mathematical models of blood coagulation and platelet adhesion: Clinical applications. Curr. Pharm. Des. 13 (14), 1457–1467.
Welsh J.D., Stalker T.J., Voronov R., Muthard R.W., Tomaiuolo M., Diamond S.L., Brass L.F. 2014. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood. 124 (11), 1808–1815.
Tomaiuolo M., Stalker T.J., Welsh J.D., Diamond S.L., Sinno T., Brass L.F. 2014. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood. 124 (11), 1816–1823.
Shattil S.J., Kim C., Ginsberg M.H. 2010. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 11, 288.
Buensuceso C., de Virgilio M., Shattil S.J. 2003. Detection of integrin alpha IIbbeta 3 clustering in living cells. J. Biol. Chem. 278 (17), 15217–15224.
Bunch T.A. 2010. Integrin alphaIIbbeta3 activation in Chinese hamster ovary cells and platelets increases clustering rather than affinity. J. Biol. Chem. 285 (3), 1841–1849.
Hato T., Pampori N., Shattil S.J. 1998. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin alphaIIb beta3. J. Cell Biol. 141 (7), 1685–1695.
Banno A., Ginsberg M.H. 2008. Integrin activation. Biochem. Soc. Trans. 36 (Pt 2), 229–234.
Böttcher R.T., Fässler R. 2014. Membrane tension drives ligand-independent integrin signaling. EMBO J. 33 (21), 2439–2442.
Shattil S.J., Newman P.J. 2004. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood. 104 (6), 1606–1615.
Moroi A.J., Watson S.P. 2015. Akt and mitogen-activated protein kinase enhance C-type lectin-like receptor 2-mediated platelet activation by inhibition of glycogen synthase kinase 3α/β. J. Thromb. Haemost. 13 (6), 1139–1150.
Lova P., Paganini S., Sinigaglia F., Balduini C., Torti M. 2002. A Gi-dependent pathway is required for activation of the small GTPase Rap1B in human platelets. J. Biol. Chem. 277 (14), 12009–12015.
Mangin P.H., Onselaer M.-B., Receveur N., Le Lay N., Hardy A.T., Wilson C., Sanchez X., Loyau S., Dupuis A., Babar A.K., Miller J.L., Philippou H., Hughes C.E., Herr A.B., Ariëns R.A., Mezzano D., Jandrot-Perrus M., Gachet C., Watson S.P. 2018. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 103 (5), 898–907.
Boylan B., Gao C., Rathore V., Gill J.C., Newman D.K., Newman P.J. 2008. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 112 (7), 2780–2786.
Antenucci L., Hytönen V.P., Ylänne J. 2018. Phosphorylated immunoreceptor tyrosine-based activation motifs and integrin cytoplasmic domains activate spleen tyrosine kinase via distinct mechanisms. J. Biol. Chem. 293 (13), 4591–4602.
Lawler J., Hynes R.O. 1989. An integrin receptor on normal and thrombasthenic platelets that binds thrombospondin. Blood. 74 (6), 2022–2027.
Rivas G.A., González-Rodríguez J. 1991. Calcium binding to human platelet integrin GPIIb/IIIa and to its constituent glycoproteins. Effects of lipids and temperature. Biochem. J. 276 (Pt 1), 35–40.
Sheldrake H.M., Patterson L.H. 2009. Function and antagonism of beta3 integrins in the development of cancer therapy. Curr. Cancer Drug Targets. 9 (4), 519–540.
Uzan G., Prenant M., Prandini M.H., Martin F., Marguerie G. 1991. Tissue-specific expression of the platelet GPIIb gene. J. Biol. Chem. 266 (14), 8932–8939.
Oki T., Kitaura J., Eto K., Lu Y., Maeda-Yamamoto M., Inagaki N., Nagai H., Yamanishi Y., Nakajima H., Kumagai H., Kitamura T. 2006. Integrin alphaIIbbeta3 induces the adhesion and activation of mast cells through interaction with fibrinogen. J. Immunol. 176 (1), 52–60.
Oki T., Eto K., Izawa K., Yamanishi Y., Inagaki N., Frampton J., Kitamura T., Kitaura J. 2009. Evidence that integrin alpha IIb beta 3-dependent interaction of mast cells with fibrinogen exacerbates chronic inflammation. J. Biol. Chem. 284 (45), 31463–31472.
Emambokus N.R., Frampton J. 2003. The glycoprotein IIb molecule is expressed on early murine hematopoietic progenitors and regulates their numbers in sites of hematopoiesis. Immunity. 19 (1), 33–45.
Nurden A.T., Pillois X., Wilcox D.A. 2013. Glanzmann thrombasthenia: State of the art and future directions. Semin. Thromb. Hemost. 39 (6), 642–655.
Di Minno G., Zotz R.B., d’Oiron R., Bindslev N., Di Minno M.N.D., Poon M.-C. 2015. The international, prospective Glanzmann Thrombasthenia Registry: Treatment modalities and outcomes of non-surgical bleeding episodes in patients with Glanzmann thrombasthenia. Haematologica. 100 (8), 1031–1037.
Tsuji S., Sugimoto M., Miyata S., Kuwahara M., Kinoshita S., Yoshioka A. 1999. Real-time analysis of mural thrombus formation in various platelet aggregation disorders: Distinct shear-dependent roles of platelet receptors and adhesive proteins under flow. Blood. 94 (3), 968–975.
Springer T.A. 1997. Folding of the N-terminal, ligand-binding region of integrin alpha-subunits into a beta-propeller domain. Proc. Natl. Acad. Sci. USA. 94 (1), 65–72.
Springer T.A. 2002. Predicted and experimental structures of integrins and beta-propellers. Curr. Opin. Struct. Biol. 12 (6), 802–813.
Xiong J.P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D.L., Joachimiak A., Goodman S.L., Arnaout M.A. 2001. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 294 (5541), 339–345.
Ma Y.-Q., Qin J., Plow E.F. 2007. Platelet integrin alpha(IIb)beta(3): Activation mechanisms. J. Thromb. Haemost. 5 (7), 1345–1352.
Campbell I.D., Humphries M.J. 2011. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 3 (3), a004994.
Zhu J., Luo B.-H., Xiao T., Zhang C., Nishida N., Springer T.A. 2008. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell. 32 (6), 849–861.
Luo B.-H., Carman C. V., Springer T.A. 2007. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647.
Calvete J.J., Henschen A., Gonzalez-Rodriguez J. 1989. Complete localization of the intrachain disulphide bonds and the N-glycosylation points in the alpha-subunit of human platelet glycoprotein IIb. Biochem. J. 261 (2), 561–568.
Xiong J.-P., Stehle T., Goodman S.L., Arnaout M.A. 2003. Integrins, cations and ligands: Making the connection. J. Thromb. Haemost. 1 (7), 1642–1654.
Xiong J.-P., Mahalingham B., Alonso J.L., Borrelli L.A., Rui X., Anand S., Hyman B.T., Rysiok T., Muller-Pompalla D., Goodman S.L., Arnaout M.A. 2009. Crystal structure of the complete integrin alphaVbeta3 ectodomain plus an alpha/beta transmembrane fragment. J. Cell Biol. 186 (4), 589–600.
Beglova N., Blacklow S.C., Takagi J., Springer T.A. 2002. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9 (4), 282–287.
Takagi J., Petre B.M., Walz T., Springer T.A. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 110 (5), 511–599.
Yang J., Ma Y.-Q., Page R.C., Misra S., Plow E.F., Qin J. 2009. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc. Natl. Acad. Sci. USA. 106 (42), 17729–17734.
Liu S., Calderwood D.A., Ginsberg M.H. 2000. Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 113 (Pt 2), 3563–3571.
Aylward K., Meade G., Ahrens I., Devocelle M., Moran N. 2006. A novel functional role for the highly conserved alpha-subunit KVGFFKR motif distinct from integrin alphaIIbbeta3 activation processes. J. Thromb. Haemost. 4 (8), 1804–1812.
Vinogradova O., Velyvis A., Velyviene A., Hu B., Haas T.A., Plow E.F., Qin J. 2002. A Structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 110 (5), 587–597.
Weljie A.M., Hwang P.M., Vogel H.J. 2002. Solution structures of the cytoplasmic tail complex from platelet integrin alpha IIb- and beta 3-subunits. Proc. Natl. Acad. Sci. USA. 99 (9), 5878–5883.
Calderwood D.A., Fujioka Y., de Pereda J.M., Garcia-Alvarez B., Nakamoto T., Margolis B., McGlade C.J., Liddington R.C., Ginsberg M.H. 2003. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. USA. 100 (5), 2272–2277.
Calderwood D.A., Zent R., Grant R., Rees D.J., Hynes R.O., Ginsberg M.H. 1999. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274 (40), 28071–28074.
Tadokoro S., Shattil S.J., Eto K., Tai V., Liddington R.C., de Pereda J.M., Ginsberg M.H., Calderwood D.A. 2003. Talin binding to integrin beta tails: A final common step in integrin activation. Science. 302 (5642), 103–106.
Martel V., Racaud-Sultan C., Dupe S., Marie C., Paulhe F., Galmiche A., Block M.R., Albiges-Rizo C. 2001. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J. Biol. Chem. 276 (24), 21217–21227.
Wegener K.L., Partridge A.W., Han J., Pickford A.R., Liddington R.C., Ginsberg M.H., Campbell I.D. 2007. Structural basis of integrin activation by talin. Cell. 128 (1), 171–182.
D’Souza M.-A.M.A., Kimble R.M., McMillan J.R. 2010. Kindler syndrome pathogenesis and fermitin family homologue 1 (kindlin-1) function. Dermatol. Clin. 28 (1), 115–118.
Moser M., Legate K.R., Zent R., Fassler R. 2009. The tail of integrins, talin, and kindlins. Science. 324 (5929), 895–899.
Kammerer P., Aretz J., Fässler R. 2017. Lucky kindlin: A cloverleaf at the integrin tail. Proc. Natl. Acad. Sci. USA. 114 (35), 9234–9236.
Brunner M., Millon-Fremillon A., Chevalier G., Nakchbandi I.A., Mosher D., Block M.R., Albiges-Rizo C., Bouvard D. 2011. Osteoblast mineralization requires beta1 integrin/ICAP-1-dependent fibronectin deposition. J. Cell Biol. 194 (2), 307–322.
Shock D.D., Naik U.P., Brittain J.E., Alahari S.K., Sondek J., Parise L.V. 1999. Calcium-dependent properties of CIB binding to the integrin alphaIIb cytoplasmic domain and translocation to the platelet cytoskeleton. Biochem. J. 342 (Pt 3), 729–735.
Larkin D., Murphy D., Reilly D.F., Cahill M., Sattler E., Harriott P., Cahill D.J., Moran N. 2004. ICln, a novel integrin alphaIIbbeta3-associated protein, functionally regulates platelet activation. J. Biol. Chem. 279 (26), 27286–27293.
Kato A., Kawamata N., Tamayose K., Egashira M., Miura R., Fujimura T., Murayama K., Oshimi K. 2002. Ancient ubiquitous protein 1 binds to the conserved membrane-proximal sequence of the cytoplasmic tail of the integrin alpha subunits that plays a crucial role in the inside-out signaling of alpha IIbbeta 3. J. Biol. Chem. 277 (32), 28934–28941.
Vijayan K.V., Liu Y., Li T.-T., Bray P.F. 2004. Protein phosphatase 1 associates with the integrin alphaIIb subunit and regulates signaling. J. Biol. Chem. 279 (32), 33039–33042.
Rantala J.K., Pouwels J., Pellinen T., Veltel S., Laasola P., Mattila E., Potter C.S., Duffy T., Sundberg J.P., Kallioniemi O., Askari J.A., Humphries M.J., Parsons M., Salmi M., Ivaska J. 2011. SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat. Cell Biol. 13 (11), 1315–1324.
Sveshnikova A.N., Balatskiy A.V., Demianova A.S., Shepelyuk T.O., Shakhidzhanov S.S., Balatskaya M.N., Pichugin A. V., Ataullakhanov F.I., Panteleev M.A. 2016. Systems biology insights into the meaning of the platelet’s dual-receptor thrombin signaling. J. Thromb. Haemost. 14 (10), 2045–2057.
Horowitz L.F., Hirdes W., Suh B.-C., Hilgemann D.W., Mackie K., Hille B. 2005. Phospholipase C in living cells: Activation, inhibition, Ca2+ requirement, and regulation of M current. J. Gen. Physiol. 126 (3), 243–262.
Shaturny V.I., Shakhidzhanov S.S., Sveshnikova A.N., Panteleev M.A. 2014. Activators, receptors and signal transduction pathways of blood platelets. Biomed. Khimiya (Rus). 60 (2), 182–200.
Burkhart J.M., Vaudel M., Gambaryan S., Radau S., Walter U., Martens L., Geiger J., Sickmann A., Zahedi R.P. 2012. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 120 (1528–0020 (Electronic)), e73–e82.
Lian L., Wang Y., Draznin J., Eslin D., Bennett J.S., Poncz M., Wu D., Abrams C.S. 2005. The relative role of PLC and PI3K in platelet activation. Blood. 106 (1), 110–117.
Tsang E., Giannetti A.M., Shaw D., Dinh M., Tse J.K.Y., Gandhi S., Ho A., Wang S., Papp E., Bradshaw J.M. 2008. Molecular mechanism of the Syk activation switch. J. Biol. Chem. 283 (47), 32650–32659.
Watson S.P., Herbert J.M.J., Pollitt A.Y. 2010. GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 8 (7), 1456–1467.
Dunster J.L., Mazet F., Fry M.J., Gibbins J.M., Tindall M.J. 2015. Regulation of early steps of GPVI signal transduction by phosphatases: A systems biology a pproach. PLoS Comput. Biol. 11 (11), 1–26.
Pasquet J.M., Gross B., Quek L., Asazuma N., Zhang W., Sommers C.L., Schweighoffer E., Tybulewicz V., Judd B., Lee J.R., Koretzky G., Love P.E., Samelson L.E., Watson S.P. 1999. LAT is required for tyrosine phosphorylation of phospholipase cgamma2 and platelet activation by the collagen receptor GPVI. Mol. Cell. Biol. 19 (12), 8326–8334.
Moroi A.J., Watson S.P. 2015. Impact of the PI3-kinase/Akt pathway on ITAM and hemITAM receptors: Haemostasis, platelet activation and antithrombotic therapy. Biochem. Pharmacol. 94 (3), 186–194.
Suzuki-Inoue K., Tulasne D., Shen Y., Bori-Sanz T., Inoue O., Jung S.M., Moroi M., Andrews R.K., Berndt M.C., Watson S.P. 2002. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 277 (24), 21561–21566.
Nieswandt B., Bergmeier W., Schulte V., Rackebrandt K., Gessner J.E., Zirngibl H. 2000. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcR gamma chain. J. Biol. Chem. 275 (31), 23998–24002.
May F., Hagedorn I., Pleines I., Bender M., Vogtle T., Eble J., Elvers M., Nieswandt B. 2009. CLEC-2 is an essential platelet activating receptor in hemostasis and thrombosis. Blood. 114 (16), 3464–3473.
Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2015. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol. BioSyst. 11 (4), 1052–1060.
Stefanini L., Bergmeier W. 2014. CalDAG-GEFI and platelet activation. Platelets. 21 (4), 239–243.
Stefanini L., Bergmeier W. 2016. RAP1-GTPase signaling and platelet function. J. Mol. Med. 94 (1), 13–19.
Stefanini L., Paul D.S., Robledo R.F., Chan E.R., Getz T.M., Campbell R.A., Kechele D.O., Casari C., Piatt R., Caron K.M., Mackman N., Weyrich A.S., Parrott M.C., Boulaftali Y., Adams M.D., Peters L.L., Bergmeier W. 2015. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Invest. 125 (4), 1419–1432.
Battram A.M., Durrant T.N., Agbani E.O., Heesom K.J., Paul D.S., Piatt R., Poole A.W., Cullen P.J., Bergmeier W., Moore S.F., Hers I. 2017. The phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) binder Rasa3 regulates phosphoinositide 3-kinase (PI3K)-dependent integrin αIIbβ3outside-in signaling. J. Biol. Chem. 292 (5), 1691–1704.
Lawrence M.B., McIntire L.V., Eskin S.G. 1987. Effect of flow on polymorphonuclear leukocyte/endothelial cell adhesion. Blood. 70 (5), 1284–1290.
Turner N.A., Sartain S.E., Hui S.-K., Moake J.L. 2015. Regulatory components of the alternative complement pathway in endothelial cell cytoplasm, factor H and factor I, are not packaged in Weibel-Palade bodies. PLoS One. 10 (3), e0121994.
Gralnick H.R., Williams S.B., McKeown L.P., Magruder L., Hansmann K., VAIL M., Parker R.I. 1991. Platelet von Willebrand factor. Mayo Clin. Proc. 66 (6), 634–640.
Yin H., Liu J., Li Z., Berndt M.C., Lowell C.A., Du X. 2008. Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood. 112 (4), 1139–1146.
Canobbio I., Balduini C., Torti M. 2004. Signalling through the platelet glycoprotein Ib-V–IX complex. Cell. Signal. 16 (12), 1329–1344.
Litvinov R.I., Farrell D.H., Weisel J.W., Bennett J.S. 2016. The platelet integrin αIIbβ3 differentially interacts with fibrin versus fibrinogen. J. Biol. Chem. 291 (15), 7858–7867.
Schaff M., Tang C., Maurer E., Bourdon C., Receveur N., Eckly A., Hechler B., Arnold C., de Arcangelis A., Nieswandt B., Denis C.V., Lefebvre O., Georges-Labouesse E., Gachet C., Lanza F., Mangin P.H. 2013. Integrin α6β1 is the main receptor for vascular laminins and plays a role in platelet adhesion, activation, and arterial thrombosis. Circulation. 128 (5), 541–552.
Wu Y., Span L.M., Nygren P., Zhu H., Moore D.T., Cheng H., Roder H., DeGrado W.F., Bennett J.S. 2015. The tyrosine kinase c-Src specifically binds to the active integrin αIIbβ3 to initiate outside-in signaling in platelets. J. Biol. Chem. 290 (25), 15825–15834.
Schmaier A.A., Zou Z., Kazlauskas A., Emert-Sedlak L., Fong K.P., Neeves K.B., Maloney S.F., Diamond S.L., Kunapuli S.P., Ware J., Brass L.F., Smithgall T.E., Saksela K., Kahn M.L. 2009. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc. Natl. Acad. Sci. USA. 106 (50), 21167–21172.
Peerschke E.I.B.I. 1995. Bound fibrinogen distribution on stimulated platelets: Examination by confocal scanning laser microscopy. Am. J. Pathol. 147, 678–687.
Obydennyy S.I., Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2016. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 14 (9), 1867–1881.
Morozova D., Demianova A., Canault M., Panteleev M., Alessi M.-C., Sveshnikova. A. 2018. Identification of platelet intracellular signalling pathways controlling the reversible and irreversible platelet aggregation. Res. Pract. Thromb. Haemost. 2 (S1), 46.
Mazurov A.V., Vinogradov D.V., Kabaeva N.V., Antonova G.N., Romanov Y.A., Vlasik T.N., Antonov A.S., Smirnov V.N. 1991. A monoclonal antibody, VM64, reacts with a 130 kDa glycoprotein common to platelets and endothelial cells: Heterogeneity in antibody binding to human aortic endothelial cells. Thromb. Haemost. 66 (4), 494—499.
Chen Y., Ju L., Jackson S., Zhu C. 2018. An intermediate state of integrin αIIbβ3 mediates platelet aggregation under disturbed flow. Res. Pract. Thromb. Haemost. 2 (S1), 43.
Plow E.F., Marguerie G.A. 1980. Participation of ADP in the binding of fibrinogen to thrombin-stimulated platelets. Blood. 56 (3), 553–555.
Plow E.F., Marguerie G.A. 1980. Induction of the fibrinogen receptor on human platelets by epinephrine and the combination of epinephrine and ADP. J. Biol. Chem. 255 (22), 10971–10977.
Shattil S.J., Hoxie J.A., Cunningham M., Brass L.F. 1985. Changes in the platelet membrane glycoprotein IIb/IIIa complex during platelet activation. J. Biol. Chem. 260 (20), 11107–11114.
Bennett J.S., Vilaire G. 1979. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J. Clin. Invest. 64 (5), 1393–1401.
Isenberg W.M., Mcever R.P., Phillips D.R., Shuman M.A., Bainton D.F. 1989. Immunogold-surface replica study of ADP-induced ligand binding and fibrinogen receptor clustering in human platelets. Am. J. Anat. 85, 142–148.
Nguyen T.-H., Palankar R., Bui V.-C., Medvedev N., Greinacher A., Delcea M. 2016. Rupture forces among human blood platelets at different degrees of activation. Sci. Rep. 6, 25402.
Weisel J.W., Litvinov R.I. 2013. Mechanisms of fibrin polymerization and clinical implications. Blood. 121 (10), 1712–1719.
Höök P., Litvinov R.I., Kim O. V., Xu S., Xu Z., Bennett J.S., Alber M.S., Weisel J.W. 2017. Strong binding of platelet integrin αIIbβ3 to fibrin clots: Potential target to destabilize thrombi. Sci. Rep. 7 (1), 13001.
Litvinov R.I., Shuman H., Bennett J.S., Weisel J.W. 2002. Binding strength and activation state of single fibrinogen-integrin pairs on living cells. Proc. Natl. Acad. Sci. USA. 99 (11), 7426–7431.
Topalov N.N., Kotova Y.N., Vasil’ev S.A., Panteleev M.A. 2012. Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 157 (1), 105–115.
Shakhidzhanov S.S., Shaturny V.I., Panteleev M.A., Sveshnikova A.N. 2015. Modulation and pre-amplification of PAR1 signaling by ADP acting via the P2Y12 receptor during platelet subpopulation formation. Biochim. Biophys. Acta. 1850 (12), 2518–2529.
Obydennyy S.I., Sveshnikova A.N., Ataullakhanov F.I., Panteleev M.A. 2016. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 14 (9), 1867–1881.
Belyaev A.V., Dunster J.L., Gibbins J.M., Panteleev M.A., Volpert V. 2018. Modeling thrombosis in silico: Frontiers, challenges, unresolved problems and milestones. Phys. Life Rev. 26–27, 57–95.
Panteleev M.A., Saenko E.L., Ananyeva N.M., Ataullakhanov F.I. 2004. Kinetics of Factor X activation by the membrane-bound complex of Factor IXa and Factor VIIIa. Biochem. J. 381 (Pt 3), 779–794.
Zakharova N.V., Artemenko E.O., Podoplelova N.A., Sveshnikova A.N., Demina I.A., Ataullakhanov F.I., Panteleev M.A. 2015. Platelet surface-associated activation and secretion-mediated inhibition of coagulation factor XII. PLoS One. 10 (2), e0116665.
Topalov N.N., Yakimenko A.O., Canault M., Artemenko E.O., Zakharova N.V., Abaeva A.A., Loosveld M., Ataullakhanov F.I., Nurden A.T., Alessi M.-C., Panteleev M.A. 2012. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin alpha(IIb)beta(3). Arterioscler. Thromb. Vasc. Biol. 32 (10), 2475–2483.
Dale G.L., Friese P., Batar P., Hamilton S.F., Reed G.L., Jackson K.W., Clemetson K.J., Alberio L. 2002. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 415 (6868), 175–179.
Munnix I.C.A., Kuijpers M.J.E., Auger J., Thomassen C.M.L.G.D., Panizzi P., van Zandvoort M.A.M., Rosing J., Bock P.E., Watson S.P., Heemskerk J.W.M. 2007. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: Regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 27 (11), 2484–2490.
Mattheij N.J.A., Gilio K., van Kruchten R., Jobe S.M., Wieschhaus A.J., Chishti A.H., Collins P., Heem-skerk J.W.M., Cosemans J.M.E.M. 2013. Dual mechanism of integrin alphaIIbbeta3 closure in procoagulant platelets. J. Biol. Chem. 288 (19), 13325–13336.
Artemenko E.O., Yakimenko A.O., Pichugin A. V., Ataullakhanov F.I., Panteleev M.A. 2016. Calpain-controlled detachment of major glycoproteins from the cytoskeleton regulates adhesive properties of activated phosphatidylserine-positive platelets. Biochem. J. 473 (4), 435–448.
Podoplelova N.A., Sveshnikova A.N., Kotova Y.N., Eckly A., Receveur N., Nechipurenko D.Y., Obydennyi S.I., Kireev I.I., Gachet C., Ataullakhanov F.I., Mangin P.H., Panteleev M.A. 2016. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. 128 (13), 1745–1755.
Podoplelova N.A., Sveshnikova A.N., Kurasawa J.H., Sarafanov A.G., Chambost H., Vasil’ev S.A., Demina I.A., Ataullakhanov F.I., Alessi M.-C., Panteleev M.A. 2016. Hysteresis-like binding of coagulation factors X/Xa to procoagulant activated platelets and phospholipids results from multistep association and membrane-dependent multimerization. Biochim. Biophys. Acta. 1858 (6), 1216–1227.
Yakimenko A.O., Verholomova F.Y., Kotova Y.N., Ataullakhanov F.I., Panteleev M.A. 2012. Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys. J. 102 (10), 2261–2269.
Abaeva A.A., Canault M., Kotova Y.N., Obydennyy S.I., Yakimenko A.O., Podoplelova N.A., Kolyadko V.N., Chambost H., Mazurov A.V., Ataullakhanov F.I., Nurden A.T., Alessi M.-C., Panteleev M.A. 2013. Procoagulant platelets form an alpha-granule protein-covered “cap” on their surface that promotes their attachment to aggregates. J. Biol. Chem. 288 (41), 29621–29632.
Yuki K., Bu W., Shimaoka M., Eckenhoff R. 2013. Volatile anesthetics, not intravenous anesthetic propofol bind to and attenuate the activation of platelet receptor integrin αIIbβ3. PLoS One. 8 (4), e60415.
Assinger A., Volf I., Schmid D. 2015. A Novel, rapid method to quantify intraplatelet calcium dynamics by ratiometric flow cytometry. PLoS One. 10 (4), 1–15.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Kaneva
Abbreviations: ADMIDAS, domain adjacent to MIDAS; Btk, Bruton tyrosine kinase; CalDAG-GEFI, Ca2+ and diacylglycerol regulated guanine nucleotide exchange factor I; CHO, Chinese hamster ovary cells; CLEC-2, C-type lectin-like receptor 2; DAG, diacylglycerol; DTS, dense tubular system; EGF, Epidermal Growth Factor; FERM, 4.1 protein, ezrin, radixin, moesin; GPCR, G-protein-coupled receptor; GPIb, glycoprotein Ib; GPVI, glycoprotein VI; ICln, chloride channel regulatory protein; IP3, inositol trisphosphate; ITAM, immunoreceptor tyrosine-based activation motif; LAT, linker for activation of T cells; LIMBS, ligand-associated metal-binding site; MIDAS, metal-ion dependent adhesion site; PAR, protease-activated receptor; PH domain, pleckstrin homology domain; PI3K, phosphatidylinositide 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKA, protein kinase A; PKC, protein kinase C; PLCβ(γ-2),Phospholipase Cβ(γ-2); PP1, protein phosphatase 1; RASA3, Ras GTPase-activating protein 3; RIAM, Rap1–GTP-interacting adapter molecule; SFK, Src family kinases; SH-2, Src homology 2 domain; Syk, Spleen tyrosine kinase; TRAP, thrombin receptor-activating peptide; VWF, von Willebrand factor.
Rights and permissions
About this article

Cite this article
Kaneva, V.N., Martyanov, A.A., Morozova, D.S. et al. Platelet Integrin αIIbβ3: Mechanisms of Activation and Clustering; Involvement into the Formation of the Thrombus Heterogeneous Structure. Biochem. Moscow Suppl. Ser. A 13, 97–110 (2019). https://doi.org/10.1134/S1990747819010033
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990747819010033