
Abstract
Every fourth person in the world currently has kidney problems to one or another degree. It is known that the novel coronavirus infection (COVID-19) is primarily a respiratory disease, but the kidney is a target organ. The coronavirus is tropic to kidney tissue due to the presence in the organ of RNA of angiotensin-converting enzyme type 2 and transmembrane serine protease 2, which is considered to be a target of this virus. The presence of renal failure in any stage is an independent unfavorable risk factor for contracting coronavirus and leads to a high frequency of hospitalization and mortality rate. Kidney failure is caused by various pathogenetic mechanisms: the direct cytopathic effect of the virus on their structures (podocytes, mesangial cells in the renal corpuscle, capillary endothelium in the glomerulus, epithelial cells in the proximal tubules), cytokine storm, damage to the renin–angiotensin–aldosterone system, and immunothrombosis. In many patients with confirmed coronavirus infection, from the first days of the disease, laboratory tests show significant changes in urine analysis (hematuria, proteinuria) and increased level of creatinine in the blood serum. The development of acute kidney injury is a main mortality risk factor. More research is needed into the exact effects of SARS-CoV-2 on the kidneys. Understanding the main pathogenetic pathways of their damage in COVID-19 is necessary to develop strategies and effective treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
An outbreak of atypical pneumonia was reported in the city of Wuhan (Hubei Province, China) in the winter of 2019. The causative agent was subsequently identified as a new human coronavirus (2019-nCoV), or SARS-CoV-2 (severe acute respiratory syndrome coronavirus). In February 2020, the WHO named this disease COVID-19 (COronaVIrus Disease 2019). Later, the outbreak became global, spreading across the world, and was declared a pandemic (Baloch et al., 2020; Habibzadeh and Lang, 2020; Muralidar et al., 2020; Mehandru and Merad, 2022). Most people infected with SARS-CoV-2 had upper respiratory tract involvement and mild to moderate acute respiratory symptoms that resolved within 6–10 days. However, almost 20% of patients developed serious complications, such as atypical bilateral pneumonia and/or acute respiratory distress syndrome, as well as severe multiorgan damage, leading to high mortality in the population (Cummings et al., 2020; Hu et al., 2021). In April 5, 2021, according to WHO data, the number of deaths from COVID-19 had reached 2.85 million out of 131 million cases. By comparison, seasonal influenza causes 250 000–500 000 deaths annually, with global mortality during the 2009 H1N1 influenza pandemic ranging from 151.7 to 575.4 thousand deaths (Johnson et al., 2022).
The main target of the virus was initially considered to be lung tissue, but it can affect other organs and systems as well. The mechanism of occurrence and development of this disease can be divided into the following stages: airborne penetration of the virus into the upper respiratory tract, interaction with the target cell and penetration into it, and virus replication and damage to cellular structures. If the reaction of the immune system is ineffective, the pathogen spreads throughout the body through the bloodstream, which results in the development of viremia and production of cytokines that determines pathological changes in organs (Khaitovich and Ermachkova, 2020).
Studies of SARS-CoV-2 genome have revealed the features of its structure, in which the important issue is the structure of the spike protein (S-protein) located on the surface of the virus. It contains an S1 subunit for binding to the cell receptor and S2 subunit for fusion with the cell membrane. The structure of the S protein imitates the angiotensin converting enzyme 2 (ACE2) (Kirtipal et al., 2020; Niu et al., 2023), and, therefore, viral particles effectively come into contact with ACE2 receptors with high binding affinity (Verano-Braga et al., 2020; Laghlam et al., 2021). In the classical endocrine model of the renin–angiotensin system (RAS), renin cleaves angiotensinogen and produces inactive peptide angiotensin-I, which is converted by the ACE to angiotensin-II. The latter mediates vasoconstriction, as well as the release of aldosterone from the adrenal glands, leading to sodium retention and increased blood pressure. The RAS also includes local systems with autocrine and paracrine effects in addition to the classical circulating RAS components with their well-known endocrine effects. It must be emphasized that the distribution and concentration of the ACE2 receptor in the body play an important role in the coronavirus route (Verano-Braga et al., 2020; Laghlam et al., 2021).
The virus is tropic to endothelial cells, and, therefore, it is capable of damaging target organs, such as the lung alveolar epithelium, gastrointestinal tract, liver, kidneys, heart, blood vessels, etc. It has been established that patients with concomitant chronic diseases have an unfavorable course and a high risk of mortality from coronavirus infection (Khaitovich and Ermachkova, 2020; Kirtipal et al., 2020).
Every year, more and more evidence is emerging indicating that coronavirus infection causes damage to the kidneys, which are considered its second target after the lungs (Li et al., 2020; Lui et al., 2020). It has been proven that the history of chronic kidney disease is a significant risk factor for the development of complications from COVID-19 and subsequent hospitalization (Oyelade et al, 2020; Wang et al., 2020; Henry and Lippi, 2020).
A significant number of patients infected with COVID-19 developed proteinuria, a urinary syndrome (7–63% of cases) (Li et al., 2020), and hematuria (20–48%) (Cheng et al., 2020; Wang et al., 2020). A common complication of coronavirus infection is acute kidney injury (AKI). The frequency of its occurrence varies significantly; among hospitalized patients, AKI was noted in 20% of cases; in the intensive care unit, in more than 50% (Nadim et al., 2020; Pei et al., 2020; Chen et al., 2020). AKI was diagnosed according to the established KDIGO (Kidney Disease Improving Global Outcomes) criteria, which include an increased creatinine level in serum of more than 0.3 mg/dL (>26.5 μmol/L) within 48 h, increased creatinine level by 1.5 times or more than the normal baseline, and a volume of urine of more than 0.5 mL/kg/h for 6 h (Khwaja, 2012). Thus, Guan et al. identified an increased creatinine level in 1.6% (12 out of 752 of patients), and Wang et al., revealed that AKI developed in 5.1% of cases (36 out of 701 patients), while temporary azotemia without the occurrence of AKI was observed in 116 patients (Wang et al., 2020; Guan et al., 2020). According to Malberti et al. (2000), during the pandemic, 82 from 2301 patients with COVID-19 admitted to the Cremona hospital were hospitalized them in the nephrology ward. The patient mortality in the nephrology department was higher than in patients with COVID-19 in other areas of the hospital during the same period (425 deaths per 1395 hospitalized patients, 30.5%), and was especially high for patients with C3-5 chronic kidney disease (88.2%). Those who died more frequently than survivors had complications, including AKI (43.2 vs. 20.0%, respectively) and sepsis (20.0 vs. 8.9%, respectively) (Malberti et al., 2020).
According to modern concepts, this disease occurs with a variety of pathogenetic mechanisms of kidney damage: intracellular activity of the pathogen itself with further cell death, excessive release of proinflammatory cytokines with the development of “cytokine storm,” features of the renin–angiotensin–aldosterone system (RAAS), hyperergic inflammation, and immune thrombosis. The action and activity of both individual and joint pathogenetic links are individual for each patient and make an equal contribution to the formation and development of kidney damage during coronavirus infection (Maltseva et al., 2021).
The purpose of this review is to analyze world literature data on the mechanisms of development of kidney damage in patients with a new coronavirus infection. When writing, the search for scientific literary sources was made using the keywords “coronavirus,” “renal injury,” and “cytokine storm” in the PubMed, Scopus, Web of Science, and eLibrary databases for the period from 2012 to 2021.
CYTOPATHIC EFFECT OF THE VIRUS
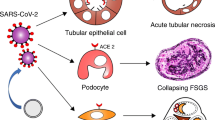
As stated above, the coronavirus penetrates into the body’s cells using ACE2 (ACE2-dependent pathway). SARS-CoV-2, entering the body, exhibits direct cytopathic effect on the renal parenchyma (Khaitovich and Ermachkova, 2020; Malik, 2020; Maltseva et al., 2021). Thus, virus fragments were observed in the urine of patients with PCR-confirmed coronavirus infection (Huang et al., 2020). When determining the primary structure of the viral RNA molecule in human tissues, it was found that the expression of ACE2 (implementation/reproduction of information) in the kidneys was almost 100 times higher than in lungs (Li et al., 2020). Most of the nephron is a target for coronavirus (Martinez-Rojas et al., 2020; Liao et al., 2020); it is expressed in the renal corpuscle (podocytes, mesangial cells), in the endothelium of capillaries of the glomerulus, and in epithelial cells of the proximal tubules (Maltseva et al., 2021). It has been found that there is 80% genetic similarity between SARS-CoV and SARS-CoV-2 (Rabaan et al., 2020).
The virus in cells triggers an element of Rip1-Rip3-MLRL (Receptor-interacting serine/threonine-protein kinase; Mixed lineage kinase domain-like pseudokinase) signaling pathway, which induces necroptosis. As a result, Rip3 oligomerizes SARS 3a and acquires the ability to integrate into the cell membrane, forming pores, and lysosomes, promoting the release of lysosomal enzymes. In turn, SARS 3a induces gene expression of transcription factor EB (TFEB) genes, which is the main regulator of lysosome biogenesis and function, lysosomal exocytosis, and autophagy (Napolitano and Ballabio, 2016; Maltseva et al., 2021). Protein E also has a cytopathic effect and is involved in the formation of ion channels in membranes of the Golgi complex that produces ionic imbalance inside the cell, activating NLRP3 inflammasome, which is accompanied by the launch of the inflammatory reaction and pyroptosis (through activation of caspase-1-dependent mechanism) (Yue et al., 2018; Maltseva et al., 2021). The interaction of the virus with auxiliary proteins allows it to penetrate inside the nephron, which leads to the death of kidney cells due to cytopathic effect (Maltseva et al., 2021).
CYTOKINE STORM
In a healthy person, there is a balance between pro- and anti-inflammatory cytokines. When a virus enters, this balance is disrupted, which leads to the development of a hyperergic inflammatory response to a foreign antigen (pathogen-associated molecular pattern (PAMP): viral RNA and viral membrane glycoprotein). PAMP is recognized through Toll-like receptors (TLRs, a type of pattern-recognition receptor (PRR)). There are different subtypes of TLR. Thus, TLR3, TLR7, TLR8, and TLR9 are involved in the detection process of SARS-CoV-2 RNA, and glycoproteins of the virus membrane are recognized by TLR2 and TLR4. During recognition of coronavirus, TLRs undergo conformational changes and complexes with participation of adapter molecules are formed that activate signaling pathways mediated by the nuclear factor κB (NF-κB), mitogen-activated protein kinases (MAPK), and interferon regulatory factors (IRF-3/5/7). As a result, under the influence of these stimuli, the production of proinflammatory cytokines, interferon-1 (IFN-1), chemokines, and adhesion molecules increase (Ragab et al., 2020; Maltseva et al., 2021).
The above-described active substances promote the infiltration of immune system cells, phagocytes, and B- and T-lymphocytes at the inflammation site. During the incubation period, the immune system does not respond to the virus for a long time; only during active replication of the coronavirus does viral PAMP accumulate, which further activates the hyperergic immune response (Paces et al., 2020; Song et al., 2020; Maltseva et al., 2021).
The development and course of a cytokine storm are systemic in nature, which results in an increased amount of proinflammatory cytokines. The latter are a direct damaging factor contributing to the development of ischemic nephropathy and microthrombosis (based on the destruction of nephrocytes and renal vessels). The proinflammatory effects of cytokines are known to have a strong influence on the development of AKI as a result of massive damage to renal tissue and impaired renal blood flow. It has been established that the most active cytokines during coronavirus infection are IL-1β, IL-6, IL-8, IL-12, IFNγ, and TNFα (Fig. 1) (Durlacher-Betzer et al., 2018; Grebe et al., 2018; Rose-John, 2018; Paces et al., 2020; Song et al., 2020; Costela-Ruiz, 2020; Maltseva et al., 2021). Thus, a large number of inflammatory mediators are involved in the pathological process and support inflammation (Grebe et al., 2018; Paces et al., 2020; Maltseva et al., 2021).

Effects of cytokines.
PATHOLOGY OF RAAS
The interaction of coronavirus with the ACE2 receptor results in RAAS disorder. ACE2 converts angiotensin II to angiotensin 1-7 and angiotensin I to angiotensin 1-9, which is then cleaved by ACE or neutral endopeptidase to produce angiotensin 1-7. The influence of angiotensin 1-7 is the opposite of the main effects of angiotensin-II (anti-inflammatory and antifibrotic). The disruption of RAAS dysregulation is that the virus interacts with ACE2 and transmembrane serine protease-2 (TMPRSS2) on the surface of nephrocytes and penetrates into the cell by endocytosis. ACE2 expression on the surface of the nephrocyte declines, which results in an enhanced content of angiotensin-II and reduced level of angiotensin 1-7 (Lelis et al., 2019; Maltseva et al., 2021) and, accordingly, to the activation of a proinflammatory effect, apoptosis, increased proliferation rate of mesangial cells, endothelial cells and fibroblasts, vasoconstriction, augmented sodium reabsorption, stimulation of aldosterone production, etc. (Lelis et al., 2019; Maltseva et al., 2021).
It has been shown that the accumulation of angiotensin-II contributes to the rapid invasion of the virus into the cells. This occurs due to the interaction of angiotensin II with a specific receptor (AT1) and cleavage of the AT1/ACE2 complex. ACE2 moves to lysosomes, and AT1 moves back to the host cell surface. The higher the level of angiotensin-II, the faster ACE2 is destroyed in lysosomes promoting accelerated penetration of SARS-CoV-2 into the cell (Maltseva et al., 2021; Ogunlade et al., 2021). It was found that accumulation of angiotensin-II enhanced the production of aldosterone by the adrenal cortex. Aldosterone, in turn, affects the generation of reactive oxygen species through an NADPH-dependent mechanism and the activity of fibroblast genes, which leads to increased collagen synthesis (Luther and Fogo, 2022). The accumulation of excess collagen is an important component in the development of fibrosis, contributing to damage to the renal parenchyma and the development of renal failure (Rafiq et al., 2011; Maltseva et al., 2021; Ogunlade et al., 2021; Luther and Fogo, 2022).
IMMUNOTHROMBOSIS
It is known that immunothrombosis is currently a major link in the pathogenesis of coronavirus infection, leading to the formation of venous and arterial blood clots and development of thrombotic complications in many organs and systems, including the kidneys. Immunothrombosis is normally necessary for protection (recognition and protection from foreign pathogen progression, since it occurs in a small area of the microvascular bed) and development of adaptive immune memory. The key role in the pathogenesis of immunothrombosis is played by immunocompetent cells (neutrophils), platelets, and the complement system (Nicolai and Massberg, 2020; Maltseva et al., 2021).
Neutrophils are a component of innate immunity. Coronavirus infection reduces ACE2 activity and promotes the release of proinflammatory molecules, which in turn are implicated in attracting neutrophils to the site of damage, as well as infiltration of damaged tissue with and induction of hyperergic inflammatory response (Nicolai and Massberg, 2020; Maltseva et al., 2021).
One of the main mechanisms that trigger and enhance immune thrombosis is the release of neutrophil extracellular traps (NETs). An NET is a three-dimensional network of decondensed chromatin, histones, and antimicrobial proteins (myeloperoxidase, elastase, pentraxin, lactoferrin, matrix metalloproteinase 9, peptidoglycan recognition protein 1) (Nicolai and Massberg, 2020; Maltseva et al., 2021). Newly formed NETs stimulate nearby platelets, thereby causing necrosis of capillary endothelial cells and renal tubular epithelial cells in AKI (Jansen et al., 2017; Nakazawa et al., 2017; Maltseva et al., 2021). Histopathological examination of kidney tissue of patients with coronavirus infection revealed in 14% patterns of neutrophils together with platelets (Schurink et al., 2020). The mechanisms of immunothrombosis formation involving NET include activation of coagulation factor XII (intrinsic coagulation pathway), binding to tissue factor and activation of the extrinsic coagulation pathway, platelet activation by histones H3 and H4, oxidation of anticoagulants (tissue factor pathway inhibitor, thrombomodulin) by neutrophil elastase and myeloperoxidase, and binding to vWF and recruitment of platelets to the site of inflammation (Jayarangaiah et al., 2020; Henry et al., 2020; Maltseva et al., 2021).
The most important role in the process of thrombus formation is given to platelets. Their function is to activate coagulation factor XII, the intrinsic coagulation pathway, and the tissue factor, which creates a predisposition to the formation of fibrin (Maltseva et al., 2021). Thus, Wang et al. found that patients with confirmed coronavirus infection had thrombocytopenia, prolongation of prothrombin and activated partial thromboplastin time, decreased fibrinogen content, increased concentration of D-dimers, and fibrin degradation products (Wang et al., 2020). A high capacity of platelets for aggregation and adhesion was noted in patients staying in the intensive care unit (Manne et al., 2020). It is taken into account that the functional activity of platelets is enhanced due to the induction of the MAPK signaling pathway and is associated with the activation of cytosolic phospholipase A2 and enhanced production and release of thromboxane A2 by platelets at the site of inflammation. It follows from this that platelets during coronavirus infection trigger a cascade of reactions of the external and internal coagulation pathways, which activates immunothrombosis in the renal vessels, initiating ischemia of the organ (Manne et al., 2020; Maltseva et al., 2021).
Another known component of the immune response to SARS-CoV-2 invasion is the complement system. However, its excessive activation and deposition of proteins result in damage of endothelium and vascular wall (Maltseva et al., 2021), as well as the development of renal vascular thrombosis and intravascular coagulation. The membrane attack complex (C5b-9) in the renal parenchyma promotes the development of acute tubular necrosis and AKI. In turn, the formed complexes C5b-9 and C5a support the release of IL-8 by endothelial cells which activates chemotaxis, adhesion and interendothelial movement of polymorphonuclear leukocytes and macrophages in the lesion site (Maltseva et al., 2021; Diao et al., 2021). The direct effect of the C5a complex on vascular endothelial cells induces exocytosis of von Willebrand factor molecules from Weibel–Palaade bodies, as well as platelet adhesion and increased coagulation cascade in kidney proximal tubules (Vinayagam and Sattu, 2020; Maltseva et al., 2021). Thus, with pathology of the complement system in patients suffering from coronavirus infection, massive thrombosis of the renal vessels comes to the fore, leading to death.
CONCLUSIONS
The kidneys are considered one of the sensitive organs affected by SARS-CoV-2. This review highlights the main pathological mechanisms of their damage. The pathophysiology of renal dysfunction is associated with specific mechanisms (direct viral entry, unbalanced activation of the RAAS, cytokine storm and thrombotic conditions), creating a vicious circle (Muralidar et al., 2020; Baloch et al., 2020; Habibzadeh and Lang, 2020; Cummings et al., 2020; Khaitovich et al., 2020; Hu et al., 2021; Maltseva et al., 2021; Mehandru and Merad, 2022).
Each individual link in pathogenesis supports and/or potentiates the development of another link in renal damage. Thus, direct penetration of coronavirus into nephrocytes triggers viral replication, which leads to mitochondrial damage and cytolysis (Khaitovich et al., 2020; Malik, 2020; Maltseva et al., 2021). Coronavirus infection activates and enhances influence of RAAS components (angiotensin II, renin, aldosterone), which contributes to the penetration of the virus into cells and generation of reactive oxygen species, resulting in destruction of the renal parenchyma and development of fibrosis (Lelis et al., 2019; Ogunlade et al., 2021; Maltseva et al., 2021; Luther and Fogo, 2022).
A key factor in creating the vicious cycle during coronavirus infection is the “recognition” of the coronavirus by immunocompetent cells. It stimulates the activity of specific signaling pathways in the body, thereby increasing the expression of proinflammatory cytokines (the “cytokine storm” phenomenon), and produces a proinflammatory environment in the kidney parenchyma, resulting in the development of renal damage (Grebe et al., 2018; Ragab et al., 2020; Song et al., 2020; Costela-Ruiz et al., 2020; Maltseva et al., 2021).
When coronavirus enters the body, the complex reaction of the immune and the hemostasis systems (immunothrombosis) limits the spread and further removal of microorganism pathogens from the bloodstream. The main role in the development of immunothrombosis is assigned to neutrophils and the NETs that they release. In turn, NETs contribute to increased thrombosis of renal vessels and implication of cells of the innate and acquired immune systems in the pathological process. Increased activity of platelets (during coronavirus infection, the activity of internal and external coagulation pathways is triggered and maintained) and proteins of the complement system, which enhance the development of thrombotic complications in kidneys, are also important for the development of immunothrombosis (Khwaja, 2012; Nicolai and Massberg, 2020).
The above requires the monitoring of kidney function in patients already with mild respiratory symptoms of coronavirus infection. Assessment and correction of renal dysfunction in early stages will improve the prognosis of this group of patients (Muralidar et al., 2020; Baloch et al., 2020; Habibzadeh and Lang, 2020; Cummings et al., 2020; Khaitovich et al., 2020; Hu et al., 2021; Maltseva et al., 2021; Mehandru and Merad, 2022).
Understanding the main pathogenetic pathways of renal parenchyma damage and AKI during coronavirus infection is important for development of treatment strategies and effective methods of therapy. Further research is needed to expand knowledge about the mechanisms of kidney damage during coronavirus infection to determine diagnostic and therapeutic approaches for patient management. It is extremely important to recognize the pathogenetic pathways and sites of coronavirus impact on renal structures for the development of new drugs (Muralidar et al., 2020; Baloch et al., 2020; Habibzadeh and Lang, 2020; Cummings et al., 2020; Haitovich et al., 2020; Hu et al., 2021; Maltseva et al., 2021; Mehandru and Merad, 2022).
REFERENCES
Baloch, S., Baloch, M.A., Zheng, T., and Pei, X., The coronavirus disease 2019 (COVID-19) pandemic, Tohoku J. Exp. Med., 2020, vol. 250, p. 271. https://doi.org/10.1620/tjem.250.271
Chen, T., Wu, D., Chen, H., Yan, W., Yang, D., Chen, G., Ma, K., Xu, D., Yu, H., Wang, H., Guo, W., Chen, J., Ding, C., Zhang, X., Huang, J., et al., Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study, BMJ, 2020, vol. 368, p. m1091. https://doi.org/10.1136/bmj.m1091
Cheng, Y., Luo, R., Wang, K., Zhang, M., Wang, Z., Dong, L., Li, J., Yao, Y., Ge, S., and Xu, G., Kidney disease is associated with in-hospital death of patients with COVID-19, Kidney Int., 2020, vol. 97, p. 829. https://doi.org/10.1016/j.kint.2020.03.005
Cicco, S., Cicco, G., Racanelli, V., and Vacca, A., Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment, Mediators Inflamm., 2020, vol. 2020, p. 7527953. https://doi.org/10.1155/2020/7527953
Costela-Ruiz, V.J., Illescas-Montes, R., Puerta-Puerta, J.M., Ruiz, C., and Melguizo-Rodríguez, L., SARS-CoV-2 infection: the role of cytokines in COVID-19 disease, Cytokine Growth Factor Rev., 2020, vol. 54, p. 62. https://doi.org/10.1016/j.cytogfr.2020.06.001
Cummings, M.J., Baldwin, M.R., Abrams, D., Jacobson, S.D., Meyer, B.J., Balough, E.M., Aaron, J.G., Claassen, J., Rabbani, L.E., Hastie, J., Beth, R., Hochman, B., Salazar-Schicchi, J, Yip, N., Brodie, D., et al., Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study, Lancet, 2020, vol. 395, p. 1763. https://doi.org/10.1016/S0140-6736(20)31189-2
Diao, B., Wang, C., Wang, R., Feng, Z., Zhang, J., Yang, H., Tan, Y., Wang, H., Wang, C., Liu, L., Liu, Y., Wang, G., Yuan, Z., Hou, X., Ren, L., et al., Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection, Nat. Commun., 2021, vol. 12, p. 2506. https://doi.org/10.1038/s41467-021-22781-1
Durlacher-Betzer, K., Hassan, A., Levi, R., Axelrod, J., Silver, J., and Naveh-Many, T., Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease, Kidney Int., 2018, vol. 94, p. 315. https://doi.org/10.1016/j.kint.2018.02.026
Grebe, A., Hoss, F. and Latz, E., NLRP3 Inflammasome and the IL-1 pathway in atherosclerosis, Circ. Res., 2018, vol. 122, p. 1722. https://doi.org/10.1161/CIRCRESAHA.118.311362
Guan, W.J., Ni, Z.Y., Hu, Y., Liang, W.H., Ou, C.Q., He, J.X., Liu, L., Shan, H., Lei, C.L., Hui, D S.C., Du, B., Li, L-j., Zeng, G., Yuen, K-Y., Chen, R., et al., China Medical Treatment Expert Group for COVID-19. Clinical characteristics of coronavirus disease 2019 in China, N. Engl. J. Med., 2020, vol. 382, p. 1708. https://doi.org/10.1056/NEJMoa2002032
Habibzadeh, F. and Lang, T., The coronavirus pandemic: “The Show Must NOT Go On,” Int. J. Occup. Environ. Med., 2020, vol. 11, p. 63. https://doi.org/10.34172/ijoem.2020.1979
Henry, B.M. and Lippi, G., Chronic kidney disease is associated with severe coronavirus disease 2019 (COVID-19) infection, Int. Urol. Nephrol., 2020, vol. 52, p. 1193. https://doi.org/10.1007/s11255-020-02451-9
Henry, B.M., Vikse, J., Benoit, S., Favaloro, E.J., and Lippi, G., Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis, Clin. Chim. Acta, 2020, vol. 507, p. 167. https://doi.org/10.1016/j.cca.2020.04.027
Hu, B., Guo, H., Zhou, P., and Shi, Z.L., Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol., 2021, vol. 19, p. 141. https://doi.org/10.1038/s41579-020-00459-7
Huang, C, Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., Yu, T., Xia, J., Wei, Y., Wu, W., Xie, X., et al., Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China, Lancet, 2020, vol. 395, p. 497. https://doi.org/10.1016/S0140-6736(20)30183-5
Jansen, M.P., Emal, D., Teske, G.J., Dessing, M.C., Florquin, S., and Roelofs, J.J., Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps, Kidney Int., 2017, vol. 91, p. 352. https://doi.org/10.1016/j.kint.2016.08.006
Jayarangaiah, A., Kariyanna, P.T., Chen, X., Jayarangaiah, A., and Kumar, A., COVID-19-associated coagulopathy: an exacerbated immunothrombosis response, Clin. Appl. Thromb. Hemost., 2020, vol. 26, p. 1076029620943293. https://doi.org/10.1177/1076029620943293
Johnson, A.G., Amin, A.B., Ali, A.R., Hoots, B., Cadwell, B.L., Arora, S., Avoundjian, T., Awofeso, A.O., Barnes, J., Bayoumi, N.S., Busen, K., Chang, C., Cima, M., Crockett, M., Cronquist, A., et al., COVID-19 incidence and death rates among unvaccinated and fully vaccinated adults with and without booster doses during periods of Delta and Omicron variant emergence—25 U.S. Jurisdictions, April 4–December 25, 2021, MMWR Morb. Mortal. Wkly Rep., 2022, vol. 71, p. 132. https://doi.org/10.15585/mmwr.mm7104e2
Khaitovich, A.B. and Ermachkova, P.A., Pathogenesis of COVID-19, Tavrich. Med.-Biol. Vestn., 2020, vol. 23, p. 113. https://doi.org/10.37279/2070-8092-2020-23-4-113-132
Khwaja, A., KDIGO clinical practice guidelines for acute kidney injury, Nephron. Clin. Pract., 2012, vol. 120, p. 179. https://doi.org/10.1159/000339789
Kirtipal, N., Bharadwaj, S., and Kang, S.G., From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses, Infect. Genet. Evol., 2020, vol. 85, p. 104502. https://doi.org/10.1016/j.meegid.2020.104502
Laghlam, D., Jozwiak, M. and Nguyen, L.S., Renin–angiotensin–aldosterone system and immunomodulation: a state-of-the-art review, Cells, 2021, vol. 10, p. 1767. https://doi.org/10.3390/cells10071767
Lelis, D.F., Freitas, D.F., Machado, A.S., Crespo, T.S., and Santos, S.H.S., Angiotensin-(1-7), adipokines and inflammation, Metabolism, 2019, vol. 95, p. 36. https://doi.org/10.1016/j.metabol.2019.03.006
Li, Z., Wu, M., Yao, J., Guo, J., Liao, X., Song, S., Li, J., Duan, G., Zhou, Y., Wu, X., Zhou, Z., Wang, T., Hu, M., Chen, X., Fu, Y., et al., Caution on kidney dysfunctions of COVID-19 patients, medRxiv, The preprint server for health sciences, 2020. https://doi.org/10.1101/2020.02.08.20021212
Liao, J., Yu, Z., Chen, Y., Bao, M., Zou, C., Zhang, H., Liu, D., Li, T., Zhang, Q., Li, J., Cheng, J., and Mo, Z., Single-cell RNA sequencing of human kidney, Sci. Data, 2020, vol. 7, p. 4. https://doi.org/10.1038/s41597-019-0351-8
Liu, Y., Yang, Y., Zhang, C., Huang, F., Wang, F., Yuan, J., Wang, Z., Li, J., Li, J., Feng, C., Zhang, Z., Wang, L., Peng, L., Chen, L., Qin, Y., et al., Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury, Sci. China Life Sci., 2020, pp. 63, 364. https://doi.org/10.1007/s11427-020-1643-8
Luther, J.M. and Fogo, A.B., The role of mineralocorticoid receptor activation in kidney inflammation and fibrosis, Kidney Int. Suppl. (2011), 2022, vol. 12, p. 63. https://doi.org/10.1016/j.kisu.2021.11.006
Malberti, F., Pecchini, P., Marchi, G., and Foramitti, M., When a nephrology ward becomes a COVID-19 ward: the Cremona experience, J. Nephrol., 2020, vol. 33, p. 625. https://doi.org/10.1007/s40620-020-00743-y
Malik, Y.A., Properties of coronavirus and SARS-CoV-2, Malays. J. Pathol., 2020, vol. 42, p. 3.
Maltseva, L.D., Vasalatii, I.M., Isaakyan, Yu.A., and Morozova, O.L., Mechanisms of acute kidney injury in COVID-19. Review, Nefrol. Dializ, 2021, vol. 23, p. 352. https://doi.org/10.28996/2618-9801-2021-3-352-365
Manne, B.K., Denorme, F., Middleton, E.A., Portier, I., Rowley, J.W., Stubben, C., Petrey, A.C., Tolley, N.D., Guo, L., Cody, M., Weyrich, A.S., Yost, C., Rondina, M.T., and Campbell, R.A., Platelet gene expression and function in patients with COVID-19, Blood, 2020, vol. 136, p. 1317. https://doi.org/10.1182/blood.2020007214
Martinez-Rojas, M.A., Vega-Vega, O., and Bobadilla, N.A., Is the kidney a target of SARS-CoV-2?, Am. J. Physiol. Renal Physiol., 2020, vol. 318, p. 1454. https://doi.org/10.1152/ajprenal.00160.2020
Mehandru, S. and Merad, M., Pathological sequelae of long-haul COVID, Nat. Immunol., 2022, vol. 23, p. 194. https://doi.org/10.1038/s41590-021-01104-y
Muralidar, S., Ambi, S.V., Sekaran, S., and Krishnan, U.M., The emergence of COVID-19 as a global pandemic: understanding the epidemiology, immune response and potential therapeutic targets of SARS-CoV-2, Biochimie, 2020, vol. 179, p. 85. https://doi.org/10.1016/j.biochi.2020.09.018
Nadim, M.K., Forni, L.G., Mehta, R.L., Connor, M.J., Jr., Liu, K.D., Ostermann, M., Rimmelé, T., Zarbock, A., Bell, S., Bihorac, A., Cantaluppi, V., Hoste, E., Husain-Syed, F., Germain, M.J., Goldstein, S.L., and Kellum, J.A., COVID-19-associated acute kidney injury: consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup, Nat. Rev. Nephrol., 2020, vol. 16, p. 747. https://doi.org/10.1038/s41581-020-00356-5
Nakazawa, D., Kumar, S.V., Marschner, J., Desai, J., Holderied, A., Rath, L., Kraft, F., Lei, Y., Fukasawa, Y., Moeckel, G.W., Angelotti, M.L., Liapis, H., and Anders, H.J., Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI, J. Am. Soc. Nephrol., 2017, vol. 28, p. 1753. https://doi.org/10.1681/ASN.2016080925
Napolitano, G. and Ballabio, A., TFEB at a glance, J. Cell. Sci., 2016, vol. 129, p. 2475. https://doi.org/10.1242/jcs.146365
Nicolai, L. and Massberg, S., Platelets as key players in inflammation and infection, Curr. Opin. Hematol., 2020, vol. 27, p. 34. https://doi.org/10.1097/MOH.0000000000000551
Niu, Z., Xu, S., Zhang, J., Zou, Z., Ren, L., Liu, X., Zhang, S., Zou, H., Hu, X., Wang, J., Zhang, L., Zhou, Y., and Song, Z., Bioinformatic analysis of the S protein of human respiratory coronavirus, Mol. Phylogenet. Evol., 2023, vol.181, p. 107704. https://doi.org/10.1016/j.ympev.2023.107704
Ogunlade, B.O., Lazartigues, E., and Filipeanu, C.M., Angiotensin type 1 receptor-dependent internalization of SARS-CoV-2 by angiotensin-converting enzyme 2, Hypertension, 2021, vol. 77, p. 42. https://doi.org/10.1161/HYPERTENSIONAHA.120.16795
Oyelade, T., Alqahtani, J., and Canciani, G., Prognosis of COVID-19 in patients with liver and kidney diseases: an early systematic review and meta-analysis, Trop. Med. Infect. Dis., 2020, vol. 5, p. 80. https://doi.org/10.3390/tropicalmed5020080
Paces, J., Strizova, Z., Smrz, D., and Cerny, J., COVID-19 and the immune system. Physiol. Res., 2020, vol. 69, p. 379. https://doi.org/10.33549/physiolres.934492
Pei, G., Zhang, Z., Peng, J., Liu, L., Zhang, C., Yu, C., Ma, Z., Huang, Y., Liu, W., Yao, Y., Zeng, R., and Xu, G., Renal involvement and early prognosis in patients with COVID-19 pneumonia, J. Am. Soc. Nephrol., 2020, vol. 31, p. 1157. https://doi.org/10.1681/ASN.2020030276
Rabaan, A.A., Al-Ahmed, S.H., Haque, S., Sah, R., Tiwari, R., Malik, Y.S., Dhama, K., Yatoo, M.I., Bonilla-Aldana, D.K., and Rodriguez-Morales, A.J., SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview, Infez. Med., 2020, vol. 28, p. 174.
Rafiq, K., Hitomi, H., Nakano, D., and Nishiyama, A., Pathophysiological roles of aldosterone and mineralocorticoid receptor in the kidney, J. Pharmacol. Sci., 2011, vol. 115, p. 1. https://doi.org/10.1254/jphs.10r07cr
Ragab, D., Salah Eldin, H., Taeimah, M., Khattab, R., and Salem, R., The COVID-19 cytokine storm; what we know so far, Front. Immunol., 2020, vol. 11, p. 1446. https://doi.org/10.3389/fimmu.2020.01446
Rose-John, S., Interleukin-6 family cytokines, Cold Spring Harb. Perspect. Biol., 2018, vol. 10, p. a028415. https://doi.org/10.1101/cshperspect.a028415
Schurink, B., Roos, E., Radonic, T., Barbe, E., Bouman, C.S.C., de Boer, H.H., de Bree, G.J., Bulle, E.B., Aronica, E.M., Florquin, S., Fronczek, J., Heunks, L.M.A., de Jong, M.D., Guo, L., du Long, R., et al., Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study, Lancet Microbe, 2020, vol. 1, p. 290. https://doi.org/10.1016/S2666-5247(20)30144-0
Song, P., Li, W., Xie, J., Hou, Y., and You, C., Cytokine storm induced by SARS-CoV-2, Clin. Chim. Acta, 2020, vol. 509, p. 280. https://doi.org/10.1016/j.cca.2020.06.017
Verano-Braga, T., Martins, A.L.V., Motta-Santos, D., Campagnole-Santos, M.J., and Santos, R.A.S., ACE2 in the renin-angiotensin system, Clin. Sci. (Lond.), 2020, vol. 134, p. 3063. https://doi.org/10.1042/CS20200478
Vinayagam, S. and Sattu, K., SARS-CoV-2 and coagulation disorders in different organs, Life Sci., 2020, vol. 260, p. 118431. https://doi.org/10.1016/j.lfs.2020.118431
Wang, L., He, W.B., Yu, X.M., Hu, D.L., and Jiang, H., Prolonged prothrombin time at admission predicts poor clinical outcome in COVID-19 patients, World J. Clin. Cases, 2020a, vol. 8, p. 4370. https://doi.org/10.12998/wjcc.v8.i19.4370
Wang, L., Li, X., Chen, H., Yan, S., Li, D., Li, Y., and Gong, Z., Coronavirus disease 19 infection does not result in acute kidney injury: an analysis of 116 hospitalized patients from Wuhan, China, Am. J. Nephrol., 2020b, vol. 51, p. 343. https://doi.org/10.1159/000507471
Wang, X., Fang, X., Cai, Z., Wu, X., Gao, X., Min, J. and Wang, F., Comorbid chronic diseases and acute organ injuries are strongly correlated with disease severity and mortality among COVID-19 patients: a systemic review and meta-analysis, Research (Wash. D.C.). 2020c, vol. 2020, p. 2402961. https://doi.org/10.34133/2020/2402961
Yue, Y., Nabar, N.R., Shi, C.S., Kamenyeva, O., Xiao, X., Hwang, I.-Y., Wang, M., and Kehrl, J.H., SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death, Cell Death Dis., 2018, vol. 9, p. 904. https://doi.org/10.1038/s41419-018-0917-y
Funding
This work was supported by ongoing institutional funding (Kemerovo State Medical University of the Russian Ministry of Health). No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This work does not contain any studies involving human and animal subjects.
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
Additional information
Translated by I. Fridlyanskaya
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Utkina, E.V., Novakovskaya, V.V., Egorova, M.V. et al. Mechanisms of Kidney Damage Development in Patients with New Coronavirus Infection: Literature Review. Cell Tiss. Biol. 18, 257–264 (2024). https://doi.org/10.1134/S1990519X24700196
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990519X24700196