
Abstract
Polyurethane is a biocompatible polymer for body tissue regeneration. To improve compatibility as a low-cost source, polyurethane was mixed with hydroxyapatite derived from chicken egg shells. The hydroxyapatite is a key mineral of bone to preserve the structure’s rigidity. The polyurethane was synthesized by prepolymer process and blended by solvent casting technique with egg shell derived hydroxyapatite. The presence of characteristic bands from FTIR spectra confirmed the synthesis of egg shell derived hydroxyapatite, polymer and composites. Egg shell derived hydroxyapatite’s crystallite size was 44 Å with a hexagonal structure. X-ray diffraction supported egg shell derived hydroxyapatite incorporation into the polyurethane matrix. Through egg shell derived hydroxyapatite applying to polyurethane, composites thermal strength has been greatly increased. Scanning electron microscopy has revealed a rough morphology of composites. In phosphate buffered saline, composites had superior biodegradability. The cytocompatible property of synthesized materials was identified in-vitro Methyltetrazolium bromide bioassay. With more egg shell derived hydroxyapatite, the cell viability of the polyurethane-egg shell derived hydroxyapatite composites improved. Such findings indicate a sustainable and economic solution for tissue regeneration to synthesize biodegradable and cytocompatible materials.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Hydroxyapatite (HA), Ca10(PO4)6(OH)2, is a white solid and the crystal structure of HA belong to hexagonal crystal system. Hydroxyapatite is considered as one of the bioactive, biocompatible and osteoconductive ingredients chemically similar to bone and mineral components of the tooth [1, 2]. Nanohydroxyapatite crystals and collagen matrix make up 65% of human bone tissues. Surface orthopaedic coating, dental implantation, bone fragment replacement, a drug carrier for the delivery of a controlled drug, bone defect repair, and as an inflammatory suppressant have all been confirmed to use HA [3, 4]. More specific HA applications have also been reported such as filler, catalyst, adsorbent, alveolar ridge reconstruction, metal implant coating and middle ear implant ceramics because its structure is porous and also has heat resistance [5, 6].
Synthetic hydroxyapatite may be prepared using simple calcium ingredients from various chemicals such as Ca(OH)2, Ca(NO3)2, CaCO3 or from natural materials such as limestone and bioinorganic materials such as bone, cockle shells, shellfish, coral or egg shell [7–11]. Brittleness and fatigue were two of HA main concerns. The use of HA in combination with other polymers such as polylactide, polyamide, polycaprolactone, etc. is one of the most efficient solutions to solve these problems. Such composites of polymer and HA have a synergistic effect in applications [12, 13].
The chicken egg shell is a good and cost-effective alternate source of HA [14]. Natural source of HA is healthy and environmentally friendly. Egg shell hydroxyapatite (EHA) was synthesized by chicken egg shells which act as a calcium precursor [15]. The calcination method decomposes and removes all impurities of organic origin and pure HA is collected [16–18].
Among the various synthetic polymer groups, PU is an exclusive polymer class. It possesses high abrasion resistance, biocompatibility, flexibility, elongation ability, and surface roughness. It is well known for being easy to work with, thermoplastic, and thermally stable. Most studies support the biocompatibility and biodegradability of PU because of its unique composition and structure [19, 20]. PU is a widely used biomaterial in regenerative medicine applications like cartilage and bone repair, orthopedics, dentistry, ureteral and cardiac stents, intravascular devices, drug delivery vehicles and meniscal reconstruction. In addition to biomedical applications, it has aerospace applications. Synthetic polymers, on the other hand, have low biochemical properties due to a lack of cell recognition sites and low hydrophilicity, making them unable to interact positively with cells or tissues.
The PU and synthetic HA have been combined extensively to create composites that have shown strong promise as biomaterials [21, 22]. However, there are gaps in the literature regarding the biodegradability and cytocompatibility of HA and PU composites made from egg shells. The high load bearing capacity and bioactivity was investigated by carbon dot decorated hydroxyapatite nanohybrid for bone tissue engineering [23]. The mechanical, thermal insulation, antimicrobial and anticorrosive properties were investigated [24, 25]. However, only physical, structural, and morphological aspects of the composites were studied, leaving the biodegradability and cytocompatibility of the composites with the human Saos-2 cell line to be determined. Furthermore, it is a green and cost-effective approach to biomaterial synthesis.
The aim of this research was to improve the properties of PU by using a low-cost biofiller EHA. Eggshell contains about 95% calcium carbonate in the form of calcite, as well as organic materials including collagen, sulfated polysaccharides and proteins. The PU as a matrix loaded with 2.5 to 5% of EHA and studied its effect on the thermal stability and crystallinity of composites. To assess the biocompatibility of composites, the Methyltetrazolium bromide (MTT) bioassay used human Saos-2 cell lines, as well as their mechanical and morphological characterization. The findings indicated that the composites may be potential biomaterial for medical use.
MATERIALS AND METHODS
Materials
Isophrone diisocyanate (IPDI), dibutylindilurate (DBTDL), poly(e-caprolactone) (PCL, 1000 g/mol), tetrahydrofuran (THF) and N,N-dimethylformamide (DMF), 1,4-butanediol (BDO) have been supplied by Sigma-Aldrich Co., USA. Chicken eggs have been bought from the local market.
Polyurethane Preparation
The prepolymer process was used to produce the PU. The mole ratio of IPDI, PCL and BDO used in the synthesis was 3 : 1 : 1. In a four-neck flask, a measured quantity of PCL (8.62 g) was placed using DMF (30 mL) in N2 atmosphere. By constant stirring, the temperature was increased to 60°C. Then, IPDI (5.74 mL) and 2–3 drops of DBTDL were added to the reaction mixture. With continuous 30 min stirring, the temperature of the reaction mixture was increased to 80°C. The viscosity was increased as a result of prepolymer synthesis. After that, a determined volume of BDO (0.77 mL) was mixed and stirred for 30 min more. Finally, viscous and transparent PUR was formed. It was then poured onto Teflon plates and dried for 24 h in a 60°C oven [26, 27].
Hydroxyapatite Preparation
The shells of the eggs were thoroughly washed with water or acetone and calcinated in the aluminium crucible gradient rise 10°C/min at 900°C for 3 h in furnace. The egg shells were ground by kitchen blender to fine powder. To make Ca(OH)2 solution, the CaO obtained by calcining eggshells was combined with the distilled water. EHA was made by using a wet chemical precipitation process to combine calcium hydroxide and phosphoric acid solutions in a Ca/P 1.67 ratio to achieve a neutral pH. This solution was stirred for 5 h at 60°C with 4000 rpm. The solution was filtered and then dried in a 60°C oven for 24 h [10, 12, 28].
Preparation of PU-EHA Composites
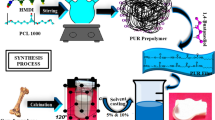
The composites of PU-EHA have been prepared using solvent casting. EHA has been incorporated into the PU matrix at two levels: 2.5 and 5% (Table 1). THF (30 mL) dissolved the prepared PU after stirring for 1 h at 60°C. Similarly, EHA was dispersed in THF (30 mL) with constant stirring at 60°C for 1 h. Then the solutions were stirred and mixed at 60°C for 3 h. After that, viscous mixture was casted onto Teflon plates and then dried at room temperature [28, 29]. The whole synthesis scheme and flow sheet of PU-EHA composites is shown in Figs. 1 and 2.

Flow sheet of synthesis of PU-EHA composites.

Practical route for preparation of PU-EHA composites.
Characterization
The elucidation of the structure of PU and PU-EHA was performed using a FTIR spectrophotometer (Agilent Technologies Carry 630 USA). With resolution of 4 cm–1 and 128 scans, the spectrum ranged from 4000 to 650 cm−1.
TGA was used to determine the thermal stability of PU and PU-EHA5 (LECO TGA-701 USA). In a N2 atmosphere (50 mL/min), the samples were heated from room temperature to 900°C. The temperature was raised at a rate of 10°C per minute.
An X-ray difractometer (Pan Analytical X’Pert Pro) was used to determine the structure of the EHA. At a scan rate of 0.01 deg/min, the pattern of X-ray diffraction was measured over a 2° range of 10°–80°. Furthermore, XRD study of the composites confirms the presence of EHA in the PU. The EHA crystallite size was determined using Scherrer formula.
TESCAN Vega LMU SEM was used to analyze the morphology of the synthesized PU and PU-EHA composites. The voltage and current supplied were 10 kV and 100 pA, respectively. For each sample, a number of images were taken at various magnifications.
The absorption of water by PU and PU-EHA composites was tested. Each sample were immersed in distilled water at room temperature for 24 h. Samples were then withdrawn, washed, and weighed again. At the time (t) the water absorption (wa) was determined by percentage formula.
Phosphate buffered saline (PBS) was used as a degradation medium to assess the biodegradability of PU and PU-EHA composites. At ambient temperature (35 ± 3°C), 0.1 g of PU and PU-EHA composites were separately dipped in the PBS media (50 mL) for 30 days. After 30 days, the PUR and composites were removed, rinsed with water. Then dried and weighed to determine loss in weight.
Saos-2 is a human osteosarcoma cell line was used for the cytotoxicity analysis of PU and PU-EHA composites. The cells were grown in Costar T-75 flasks in the Department of Microbiology and Molecular Genetics (MMG, University of the Punjab, Pakistan). Then subculturing twice a week at 37oC in a % CO2 and 100% relative humidity incubator supplied and maintained at low passage 5 to 20.
During logarithmic growth process, adherent cells had been washed with 2 mL of PBS. After that, removed by adding 1X trypsin (0.5 mL) and incubated at 37°C in an incubator for 2–5 min. 100 μL full growth media was applied to each well of microplates. Cells were calculated using Trypan blue dye staining and a hemocytometer to achieve the desired densities. At densities of 1 × 104 cells per well, each well was inoculated. The cells were given various actinomycete concentrations (25, 50, 100, and 200 μg/mL). To avoid any mistakes, all triplicate experiments are carried out. Control wells having the same volume of cell culture were used in every experiment, with the positive (Triton X-100) and negative controls. Then, the plates were incubated in CO2 at 37°C for 24 h.
After 24 h, MTT bioassay was applied to each well’s culture media. The plate was then incubated at 37°C in a 5% CO2 incubator. After 4 h, the plate was taken out from incubator without disturbing the monolayers of cells and the growth media was removed. After that, 100μL of DMSO was applied to all well and then dissolve the formazan. The spectrophotometer measured the absorbance at 570 nm. The rate of inhibition was measured and plotted for all extracts to determine their anticancer activities [27, 30].
Means were determined for quantitative analysis and multiple comparisons using an independent sample t test and two-way ANOVA were used to assess significant difference (p < 0.05).
RESULTS AND DISCUSSION
The FTIR analysis was used to monitor the entire synthesis process. Figure 3 displays spectra of EHA, monomers, PU, and PU-EHA composites. With characteristic bands of O–H (stretching vibration), carbonate, and phosphate at 3500, 1427 and 1020 cm–1, respectively, Fig. 3a confirmed the synthesis of EHA. IPDI (stretching vibration of –NCO and C–H), PCL (asymmetric and symmetric stretching of C–H hydroxyl group), and BDO (stretching vibration of ‒OH) are shown in Fig. 3B at 2940 and 2200 cm–1, 2940, 2860 cm–1 and 3200 cm–1, respectively. Figure 3c shows the spectra of PU and composites. The presence of characteristic –NH and –C=O bands of urethane and disappearance of –OH and –NCO bands of monomers in these spectra verified the synthesis of PU. In composites, the stretching vibration of –NH appeared at 3360 cm–1 in PU, with a red shift of 2 cm–1. At the lower end of the urethane –C=O stretching vibration band, there was a red shift from 1719 cm–1 (PU) to 1728 cm–1 (PU-EHA composites). The hydrogen bonding of EHA nanoparticles with the PU matrix may be responsible for these changes. Furthermore, the spectra of PU-EHA composites showed distinct carbonate and phosphate bands at 1457 and at 1032 cm–1, respectively. The results supported the proposed synthesis of PU and its composites [24–27].

FTIR spectra of (a) EHA; (b) monomers (1) IPDI, (2) PCL1000, (3) BDO; (c) composites (1) PU, (2) PU2.5 (PU with 2.5% EHA) and (3) PU5 (PU with 5% EHA).
TGA was used to examine and compare the thermal stability of PU and PU5. Figure 4 shows the corresponding thermograms. Both samples showed two stages of degradation with a similar degradation pattern. The breakdown of urethane linkages caused the highest degradation rate that was noticed between 336°C and 416°C. Because of ester bond decomposition in the polyol of PU and PU5, second stage degradation was reported around 478°C. Table 2 summarises the thermal degradation temperatures and residual weight (%) of PU and PU5. With the addition of EHA, this data clearly demonstrated some improvement in thermal stability. It could be attributed to the EHA and PU matrix’s mutual interactions. Furthermore, the residual weight (%) verified that EHA was added to the composite [20–24].

TGA thermograms of (1) PU and (2) PU5 (PU with 5% EHA).
The X-ray diffraction patterns of EHA, PU, PU2.5, and PU5 are shown in Fig. 5. EHA characteristic peaks are at 18.1° (110), 21.9° (200), 23.9° (111), 25.9° (002), 29.1° (210), 32° (211), 36.1° (301), 39.9° (310), 43.9° (113), 46.8° (222) and 48.5° (312) of 2θ. Table 3 demonstrates how these peaks are compared to JCPDS card 9-432. Furthermore, this standard data confirmed EHA’s hexagonal crystal structure. Scherrer’s formula calculated EHA’s crystallite size to be 44 Å. The amorphous diffraction pattern of PU is shown in Fig. 5b. The composites have a broad peak from 15° to 25° of 2θ in their diffraction pattern. An amorphous structure with some ordered moieties is revealed by this pattern. In addition, the PU composites revealed a range of EHA characteristic peaks, including (hkl) 002, 210, 211, 222, 310, 113, and 312, though at a lower intensity. These findings backed up a smooth and consistent integration of EHA into the PU matrix. It was most likely caused by secondary interactions between PU chains and EHA [31, 32].

XRD patterns of (a) EHA, (b) PU and composites: (1) PU, (2) PU2.5, and (3) PU 5.
SEM was used to examine the morphological properties of PU and composites. Figure 6 shows the micrographs of PU, PU2.5 and PU5. Micrographs showed that a highly porous network structure was composed of biocompatible composites. Agglomerates had irregular shape of rod and spherical form. Nano sized EHA had potential to create more beneficial porous surface. With the addition of EHA, a smooth morphology of pure PU became an extremely rough surface. Composites with rough morphologies may be suitable for use as biomaterials [10, 29].

Scanning electron micrographs of (a) PU, (b) PU2.5, and (c) PU5 at 2 µm.
MTT bioassay was used to determine the cytotoxicity of PU composites in vitro. Figure 7 shows the comparison of the results to the control. These findings were statistically analyzed using a two-way ANOVA, with a significance level of p < 0.05. These findings showed that the cell lines Saos-2 were nontoxic by the synthesized polymer and composites.

Cell viability (%) against PU, PU2.5 (composite with 2.5% EHA) and PU5 (composite with 5% EHA).
Furthermore, the addition of EHA to PU increased cell viability (%) significantly. With an increased amount of EHA in composites, an increasing trend in cell viability (%) was observed, suggesting that the commendatory addition of EHA was justified [1, 13].
Figure 8 shows the effects of testing of absorption of water of PU and PU-EHA by immersing them in distilled H2O. Both of the samples water absorption level increased over time. All immersed samples attained optimum water saturation after 24 h. The PU had the highest water absorption of 10%, while PU-EHA5 had the lowest water absorption of 8%. It may be because composites have less hydrophilic polymeric matrix than pure PU [27, 33]. Figure 8 also shows the weight loss as a measure of biodegradation of PU and PU-EHA composites in PBS. Due to hydrolyzing of soft segments, the PU was biodegradable up to 11%. Due to the fall off of EHA from the composite inside PBS, the PU5 had high weight loss of 14%. The loss of EHA caused multiple voids in the composite, weakens the PU skeleton and exposed more surfaces to hydrolytic reactions [19, 33].

(1) Biodegradability and (2) water absorption of PU, PU2.5 (PU with 2.5% EHA) and PU5 (PU with 5% EHA).
CONCLUSIONS
PU was combined with HA derived from the waste chicken egg shells. Egg shells were calcined at a high temperature to create the EHA. It was used at two separate levels in the PU matrix, 2.5 and 5%. The crystallite size of EHA was 44 Å and XRD verified that it had a hexagonal shape. The integration of EHA into the PU matrix was also supported by XRD. The synthesis was confirmed by the appearance of characteristic bands in the FTIR spectra of PU and its composites. The TGA revealed that increasing the amount of EHA in a composite improves its thermal stability. The composites’ rough morphology was shown by SEM analysis. In terms of weight loss, phosphate buffered saline had a greater biodegradability of composites. The cytocompatibility of prepared materials was determined using an in vitro MTT bioassay. Composites on the other hand, showed increased cell viability as the EHA content increased. As a result, these composites can be suggested as a biodegradable and biocompatible candidate for biomedical applications, as well as a cost effective and environmentally friendly method of preparation.
REFERENCES
I. Oladele, O. Agbabiaka, O. Olasunkanmi, A. O. Balogun, and M. O. Popoola, J. Appl. Biotechnol. Bioeng. 5, 92 (2018).
S. Sasikumar and R. Vijayaraghavan, Trends Biomater. Artif. Organs 19, 70 (2006).
T. Viana, S. Biscaia, H. A. Almeida, and P. J. Bártolo, in Proceedings of the ASME 2014 12th Biennial Conference on Engineering Systems Design and Analysis, Copenhagen, Denmark, 2014 (Copenhagen, 2014), ESDA2014-20213.
D. Zhang, W. Liu, X. D. Wu, X. He, X. Lin, H. Wang, and W. Huang, J. Mater. Sci.: Mater. Med. 30, 59 (2019).
N. I. Tkacheva, S. V. Morozov, I. A. Grigor’ev, D. M. Mognonov, and N. A. Kolchanov, Polym. Sci., Ser. B 55, 409 (2013).
G. S. Kumar, A. Thamizhavel and E. K. Girija, Mater. Lett. 76, 198 (2012).
D. S. R. Krishna, A. Siddharthan, S. K. Seshadri, and T. S. Kumar, J. Mater. Sci.: Mater. Med. 18, 1735 (2007).
D. L. Goloshchapov, V. M. Kashkarov, N. A. Rumyantseva, P. V. Seredin, A. S. Lenshin, B. L. Agapov, and E. P. Domashevskaya, Ceram. Int. 39, 4539 (2013).
E. M. Rivera, M. Araiza, W. Brostow, V. M. Castano, J. R. Dıaz-Estrada, R. Hernández, and J. R. Rodrıguez, Mater. Lett. 41, 128 (1999).
A. R. Yasmin and D. Kalyania, Int. J. Res. Appl. Sci. Eng. Technol. 3, 471 (2015).
B. Ashok, S. Naresh, K. O. Reddy, K. Madhukar, J. Cai, L. Zhang, and A. V. Rajulu, Int. J. Polym. Anal. Charact. 19, 245 (2014).
J. Yu, Y. Xu, S. Li, G. V. Seifert, M. L. Becker, Biomacromolecules. 18, 4171 (2017).
H. D. Jirimali, B. C. Chaudhari, J. C. Khanderay, S. A. Joshi, V. Singh, A. M. Patil, and V. V. Gite, Polym.-Plast. Technol. Eng. 57, 804 (2018).
P. Kamalanathan, S. Ramesh, L. T. Bang, A. Niakan, C. Y. Tan, J. Purbolaksono and W. D. Teng, Ceram. Int. 40, 16349 (2014).
R. Bardhan, S. Mahata and B. Mondal, Adv. Appl. Ceram. 110, 80 (2011).
M. M. Rahman, A. N. Netravali, B. J. Tiimob, V. Apalangya and V. K. Rangari, J. Appl. Polym. Sci. 133, 43477 (2016).
G. Gergely, F. Wéber, I. Lukács, A. L. Tóth, Z. E. Horváth, J. Mihály and C. Balázsi, Ceram. Int. 36, 803 (2010).
I. Subuki, N. Adnan and R. W. Sharudin, AIP Conf. Proc. 020019,1 (2018).
M. Kurańska, K. Polaczek, M. Auguścik-Królikowska, A. Prociak and J. Ryszkowska, Polymer 190, 122164 (2020).
Y. N . Zhao, L. Lei, D. X. Ji, and Y. H. Niu, Polym. Sci., Ser. B. 61, 595 (2019).
B. Aghajani, E. Karamian, and B. Hosseini, Nanomed. J. 4, 254 (2017).
B. C. Ang, N. Ahmad, Z. C. Ong, S. C. Cheok, and H. F. Chan, Pigm. Resin Technol. 45, 313 (2016).
C. K. Patil, H. D. Jirimali, J. S. Paradeshi, B. L. Chaudhari, and V. V. Gite, Prog. Org. Coat. 128, 127 (2019).
W. Zhao, B. Nolan, H. Bermudez, S. L. Hsu, U. Choudhary, and J. van Walsem, Polymer 193, 122344 (2020).
L. Lin, J. Shao and J. Ma, Mater. Lett. 253, 86 (2019).
I. M. Davletbaeva, S. E. Dulmaev, O. O. Sazonov, A. M. Gumerov, R. S. Davletbaev, L. R. Valiullin, and R. G. Ibragimov, Polym. Sci., Ser. B. 62, 375 (2020).
L. P. Gabriel, M. E. M dos Santos, and A. L. Jardini, Nanomed. Nanotechnol. 13, 201 (2017).
K. N. Raftopoulos, I. Łukaszewska, P. A. Klonos, E. Hebda, A. Bukowczan, A. Kyritsis, and K. Pielichowski, Polymer 182, 121821 (2019).
S. Patel, N. Gheewala and A. Suthar, Int. J. Pharm. Pharm. Sci. 1, 38 (2009).
L. Lin, J. Shao, J. Ma, Q. Zou, J. Li, Y. Zuo, and Y. Li, Mater. Lett. 253, 86 (2019).
F. Heidari, M. Razavi, M. Ghaedi, M. Forooghi, M. Tahriri, and L. Tayebi, J. Alloys Compd. 693, 1150 (2017).
S. Teixeira, M. A. Rodriguez, P. Pena, A. H. De Aza, S. De Aza, M. P. Ferraz, and F. J. Monteiro, Mater. Sci. Eng., C. 29, 1510 (2009).
Z. Cui, Z. Zheng, C. Su, J. Si, Q. Wang, and W. Chen, J. Mater. Sci. 54, 11231 (2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Shaista Parveen, Shagufta Parveen Synthesis and Characterization of Biodegradable and Cytocompatible Polyurethane-Egg Shell Derived Hydroxyapatite Biomaterials. Polym. Sci. Ser. B 63, 933–941 (2021). https://doi.org/10.1134/S1560090421060221
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1560090421060221