
Abstract
A novel heterogeneous catalyst, 1,3,5-triazine-2,4,6-triaminium trifluoromethanesulfonate (TTTMS) has been prepared, characterized, and used to catalyze the synthesis of various xanthenes via one-pot multicomponent reaction of aromatic aldehydes and dimedone or/and 2-naphthol under solvent free conditions. The proposed procedure offers several advantages, such as easy preparation of the catalyst, simple and easy workup, low cost, short reaction time, excellent yields, and mild reaction conditions. Furthermore, TTTMS is an eco-friendly, inexpensive, biodegradable, and recyclable catalyst. It can be reused four times without significant loss of catalytic activity.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Solid acid catalysts have many advantages over liquid acids in organic chemistry. Heterogeneous acidic catalysts are non-corrosive and less harmful to the environment, and they do not cause disposal of effluent problems. Furthermore, they can be easily separated from the products and are reusable. The synthesis and application of solid acid catalysts as ecologically benign and economic have attracted much attention in chemical research and industry. A large number of industrial processes utilizing heterogeneous acid catalysts have been developed by the end of the last century [1, 2]. The replacement of traditional homogeneous catalysts with heterogeneous acids is becoming an inevitable trend. Xanthenes and their derivatives are known as an important class of pharmaceutically relevant compounds since they possess different biological activities such as antibacterial [3], anti-inflammatory [4], antiviral [5], antitumor, and anticancer [6]. Additionally, xanthenes can be used in laser technology, as fluorescent materials for visualization of biomolecules, and as dyes [7]. Thus, the synthesis of these heterocyclic compounds is currently of great importance.
There are two ways to prepare symmetrical and unsymmetrical xanthenes. Symmetrical xanthenes can be obtained by the one-pot reaction of aldehydes with 2-naphthol or cyclic 1,3-dicarbonyl compounds, whereas the one-pot multicomponent reaction of aldehydes with cyclic 1,3-dicarbonyl compounds and 2-naphthol can produce unsymmetrical xanthenes. Various procedures have been introduced for the preparation of these important heterocyclics, among which the most important is cyclodehydration of aldehydes and β-naphthol and/or cyclic 1,3-dicarbonyl compounds in the presence of different Lewis and Brønsted acid catalysts such as sulfamic acid [8], silica-bonded S-sulfonic acid (SBSSA) [9], 1-butyl-3-methylimidazolium hydrogen sulfate [bmim][HSO4] [10], poly(4-vinylpyridinium)hydrogen sulfate [11], silica sulfuric acid [12], dodecatungstophosphoric acid [13], cellulose sulfonic acid [14], and nano-TiO2 [15]. Of course, most of these methods have one or more disadvantages such as long reaction time, low yield, use of toxic organic solvents, harsh reaction conditions, difficulty in the catalyst preparation, large amount of the catalysts, and tedious workup. Thus, efforts to develop alternative methods for the synthesis of xanthene derivatives are an inevitable demand.
RESULTS AND DISCUSSION
In continuation of previous studies [16, 17], the present article reports the preparation of 1,3,5-triazine-2,4,6-triaminium tris(trifluoromethanesulfonate) (TTTMS) by reaction of trifluoromethanesulfonic acid with melamine at room temperature (Scheme 1). The structure of the synthesized compound was established by IR, 1H and 13C NMR, and mass spectra, as well as by thermogravimetric and elemental analyses.

1.
The FT-IR spectra of melamine and TTTMS are shown in Fig. 1. The FT-IR spectrum of the catalyst displayed three sharp peaks at 3440, 3397, and 3230 cm–1, which can be assigned to N–H stretching of the NH3 groups. Moreover, the two peaks observed at 1393 and 1273 cm–1 correspond to symmetric and asymmetric stretching vibrations of the SO3– groups, respectively, and the band at 1238 cm–1 was assigned to SO2 stretching. In addition, C=N and C–F vibrations were observed at 1664, 1522, and 1175 cm–1, respectively.

FT-IR spectra of (1) melamine and (2) TTTMS.
The 1H NMR spectrum of TTTMS shows the NH3+ peak at δ 7.71 ppm. In the 13C NMR spectrum of TTTMS, the CF3 carbon appeared as a quartet at δC 121.1 ppm. Also, the triazine carbons appeared gave a signal at δC 159.8 ppm. The mass spectrum of 1,3,5-triazine-2,4,6-triaminium trifluoromethanesulfonate contained the expected molecular ion peak at m/z 575 together with other ion peaks at m/z 576 [M + 1]+, 574 [M – H]+, 573 [M – 2H]+, 572 [M – 3H]+, 425 [M – CF3SO3H], 276 [M – 2CF3SO3H], and 126 [M – 3CF3SO3H]+ (melamine).
Figure 2 shows the TGA diagrams of TTTMS which indicated a weight loss at <120°C related to removal of physically adsorbed water. The substrate underwent significant degradation in the temperature range 190–210°C via loss of one CF3SO3 group. The third weight loss started at 260°C (Tmax = 272°C) as a result of thermal decomposition of the second acidic group. The TGA diagrams also showed the fourth and fifth weight losses at 430–450°C (Tmax = 440°C) and 490–510°C (Tmax = 500°C). Thus, the catalyst is stable up to 210°C.

TGA curves for TTTMS.
The utility of TTTMS in organic synthesis was studied. In particular, TTTMS was used to catalyze the synthesis of xanthene derivatives via one-pot condensation of aldehydes 1a–1j with dimedone (2) and/or β-naphthol (3) under solvent-free conditions (Scheme 2). To optimize the conditions, the condensation of 4-cholorobenzaldehyde (1 mmol) and dimedone (2 mmol) was carried out in the presence of different amounts of TTTMS under solvent-free conditions at various temperatures (Table 1).

2.
The model reaction was not complete in the absence of a catalyst at room temperature nor at 90°C even after prolonging the reaction time. The obtained results indicated that the catalyst is absolutely necessary for the reaction (Table 1; entry nos. 1, 2). The best result was obtained using 5 mol % of the catalyst under solvent-free conditions at 100°C. In this case, the maximum yield of 9-(4-chlorophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (5b) was reached in the shortest reaction time (entry no. 5). The reaction with lower amount of the catalyst or at lower temperature led to a longer time and lower yield, whereas increase of the catalyst amount or rise in temperature almost did not affect the yield or reaction time (entry nos. 4, 6).
After optimization of the reaction condition, the scope of the proposed procedure was studied using differently substituted aromatic aldehydes (Table 2). As presented in Table 2, aromatic aldehydes containing electron-donating or electron-withdrawing substituents reacted with dimedone or 2-naphthol under the selected conditions in short reaction times to produce the corresponding xanthenes 4a–4j and 5a–5j in excellent yields ranging from 91 to 98% (Table 2). The catalyst was also used in the synthesis of unsymmetrical xanthene derivatives by reacting aromatic aldehydes with equimolar amounts of 2-naphthol and dimedone. The target products 6a–6j were obtained under 100°C with high yields in very short times (Table 2).
The efficiency of TTTMS in the synthesis of xanthenes in terms of reaction time, yield, and conditions was compared with other catalysts (Table 3). It is clear that the present method is more efficient, simpler, and less time consuming for the preparation of 9-(4-chlorophenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione.
Scheme 3 shows a probable reaction mechanism. First, aldehyde 1 is activated by the proton from TTTMS. The aldehyde carbonyl is then attacked by nucleophilic dimedone (2) or 2-naphthol (3) to form the Knoevenagel intermediate. Next follows nucleophilic addition of the second molecule of 2 or 3 to the Knoevenagel intermediate, and the latter undergoes intramolecular cyclization to afford final xanthene derivative 4–6.

3.
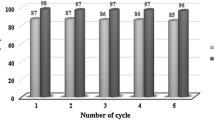
The recyclability of the catalyst in the synthesis of xanthene derivatives was investigated in the same model reaction of dimedone (2 mmol) and 4-chlorobenzaldehyde (1 mmol) under the optimized conditions. When the reaction was complete, anhydrous ethanol was added to the reaction mixture, and the catalyst was separated by filtration and reused for the next run. It was found that the catalyst can be recycled for at least four times without significant loss in yield and reaction time. The results are illustrated by Fig. 3.

Reusability of TTTMS in the reaction of dimedone and 4-chlorobenzaldehyde.
EXPERIMENTAL
All chemicals were purchased from Merck or Fluka and were used without further purification. The products were identified by comparison of their physical constants and spectral data with those of authentic samples. The melting points were determined on an electrothermal capillary melting points apparatus. The IR spectra were recorded on a Shimadzu IR-470 FT-IR spectrometer using KBr disks. The 1H and 13C NMR spectra were recorded on a Bruker DRX 400 MHz spectrometer using DMSO-d6 as solvent and reference. Elemental analyses were obtained with a Carlo Erba EA1110 CHNO-S analyzer and were in agreement with the calculated values.
1,3,5-Triazine-2,4,6-triaminium trifluoromethanesulfonate (TTTMS). A round-bottom flask (10 mL) was charged with 1,3,5-triazine-2,4,6-triamine (0.126 g, 1 mmol) and anhydrous methylene chloride (3 mL), and trifluoromethanesulfonic acid (450 mL, 3 mmol) was added dropwise over a period of 30 min at 0–5°C. The mixture was stirred at room temperature for 2 h and left to stand for 5 min, and the solvent was removed by decanting. The residue was washed with anhydrous diethyl ether (3–10 mL) and dried under reduced pressure. Yield 53.5 g (93%), white solid, mp 151–152°C. IR spectrum, ν, cm–1: 3440, 3397, 3230 (N–H), 1664, 1522 (C=N), 1393, 1273, 1238 (SO2), 1175 (C–F), 1037 (C–N). 1H NMR spectrum: δ 7.71 ppm, s (9H, NH3). 13C NMR spectrum, δC, ppm: 121.0 q (CF3, J = 322.0 Hz), 159.8 (C=N). Mass spectrum, m/z: 575 [M]+, 574 [M – H]+, 573 [M – 2H]+, 572 [M – 3H]+, 425 [M – CF3SO3H]+, 276 [M – 2CF3SO3H]+, 126 [M – 3CF3SO3H]+. Found, %: C 12.53; H 1.55; N 29.663. C6H9F9N6O9S3. Calculated, %: C 12.50; H 1.57; N 29.67.
General procedure for the preparation of xanthene derivatives. A mixture of aromatic aldehyde 1a–1j (1 mmol), dimedone (2) and/or 2-naphthol (3) (2 mmol), and TTTMS (5 mol %, 0.028 g) was stirred at 100°C under solvent-free conditions for a time indicated in Table 2. The progress of the reaction was monitored by TLC (EtOAc–n-hexane, 2:5). After completion of the reaction, the mixture was cooled and treated with anhydrous ethanol (10 mL), and the catalyst was filtered off. The filtrate was evaporated, and the residue was recrystallized from 96% ethanol.
REFERENCES
Fauziyah, M., Widiyastuti, W., and Setyawan, H., Adv. Powder Technol., 2020, vol. 31, p. 1412. https://doi.org/10.1016/j.apt.2020.01.022
Zhang, B., Gao, M., Geng, J., Cheng, Y., Wang, X., Wu, C., Wang, Q., Liu, S., and Cheung, S.M., Renewable Energy, 2021, vol. 164, p. 824. https://doi.org/10.1016/j.renene.2020.09.076
Khakia, D., Namazi, H., and Amininasab, S.M., React. Funct. Polym., 2021, vol. 158, article ID 104780. https://doi.org/10.1016/j.reactfunctpolym.2020.104780
Banerjee, A.G., Kothapalli, L.P., Sharma, P.A., Thomas, A.B., Nanda, R.K., Shrivastava, S.K., and Khatanglekar, V.V., Arab. J. Chem., 2016, vol. 9, p. S480. https://doi.org/10.1016/j.arabjc.2011.06.001
Abdel-Lateef, M.A., Omar, M.A., Ali, R., and Derayea, S.M., Microchem. J., 2019, vol. 145, p. 672. https://doi.org/10.1016/j.microc.2018.11.038
Catanzaro, E., Seghetti, F., Calcabrini, C., Rampa, A., Gobbi, S, Sestili, P., Turrini, E, Maffei, F., Hrelia, P., Bisi, A., Belluti, F., and Fimognari, C., Bioorg. Chem., 2019, vol. 86, p. 538. https://doi.org/10.1016/j.bioorg.2019.02.017
Romieu, A., Dejouy, G., and Valverde, I.E., Tetrahedron Lett., 2018, vol. 59, p. 4574. https://doi.org/10.1016/j.tetlet.2018.11.031
Rajitha, B., Kumar, B.S., Reddy, Y.T., Reddy, P.N., and Sreenivasulu, N., Tetrahedron Lett., 2005, vol. 46, p. 8691. https://doi.org/10.1016/j.tetlet.2005.10.057
Niknam, K., Panahi, F., Saberi, D., and Mohagheghnejad, M., J. Heterocycl. Chem., 2010, vol. 47, p. 292. https://doi.org/10.1002/jhet.303
Niknam, K. and Damya, M., J. Chin. Chem. Soc., 2009, vol. 56, p. 659. https://doi.org/10.1002/jccs.200900098
Khaligh, N.G., Ultrason. Sonochem., 2012, vol. 19, p. 736. https://doi.org/10.1016/j.ultsonch.2011.12.004
Seyyedhamzeh, M., Mirzaei, P., and Bazgir, A., Dyes Pigm., 2008, vol. 76, p. 836. https://doi.org/10.1016/j.dyepig.2007.02.001
Wang, H.J., Ren, X.Q., Zhang, Y.Y., and Zhang, Z.H., J. Braz. Chem. Soc., 2009, vol. 20, p. 1939. https://doi.org/10.1590/S0103-50532009001000025
Oskooie, H.A., Tahershamsi, L., Heravi, M.M., and Baghernejad, B., Eur. J. Chem., 2010, vol. 7, p. 717. https://doi.org/10.1155/2010/936107
Mirjalili, B.F., Bamoniri, A., Akbari, A., and Taghavinia, N., J. Iran. Chem. Soc., 2011, vol. 8, p. S129. https://doi.org/10.1007/BF03254289
Mohammadi Zeydi, M., Montazeri, N., and Fouladi, M., J. Heterocycl. Chem., 2017, vol. 54, p. 3549. https://doi.org/10.1002/jhet.2979
Mohammadi Zeydi, M. and Mahmoodi, N.O., J. Chin. Chem. Soc., 2017, vol. 64, p. 1023. https://doi.org/10.1002/jccs.201700069
Li, J.J., Tao, X.Y., and Zhang, Z.H., Phosphorus, Sulfur Silicon Relat. Elem., 2008, vol. 183, p. 1672. https://doi.org/10.1080/10426500701724530
Venkatesan, K., Pujari, S., Lahoti, R.J., and Srinivasan, K.V., Ultrason. Sonochem., 2008, vol. 15, p. 548. https://doi.org/10.1016/j.ultsonch.2007.06.001
Dabiri, M., Baghbanzadeh, M., Nikcheh, M.S., and Arzroomchilar, E., Bioorg. Med. Chem. Lett., 2008, vol. 18, p. 436. https://doi.org/10.1016/j.bmcl.2007.07.008
Baghbanian, S., Khanzad, G., Vahdat, S., and Tashakkorian, H., Res. Chem. Intermed., 2015, vol. 41, p. 9951. https://doi.org/10.1007/s11164-015-2001-x
Zolfigol, M.A., Khakyzadeh, V., Moosavi-Zare, A.R., Zare, A., Azimi, S.B., Asgari, Z., and Hasaninejad, A., C. R. Chim., 2012, vol. 15, p. 719. https://doi.org/10.1016/j.crci.2012.05.003
Rostamizadeh, S., Shadjou, N., Amani, A.M., and Balalaie, S., Chin. Chem. Lett., 2008, vol. 19, p. 1151. https://doi.org/10.1016/j.cclet.2008.07.026
Maleki, B., Barzegar, S., Sepehr, Z., Kermanian, M., and Tayebee, R., J. Iran. Chem. Soc., 2012, vol. 9, p. 757. https://doi.org/10.1007/s13738-012-0092-5
Abdollahi-Alibeik, M. and Ahmadi, G., Res. Chem. Intermed., 2015, vol. 41, p. 8173. https://doi.org/10.1007/s11164-014-1883-3
Zhou, Z. and Deng, X., J. Mol. Catal. A: Chem., 2013, vol. 367, p. 99. https://doi.org/10.1016/j.molcata.2012.11.002
Rad-Moghadam, K. and Azimi, S.C., J. Mol. Catal. A: Chem., 2012, vol. 363, p. 465. https://doi.org/10.1016/j.molcata.2012.07.026
Li, J.J., Tao, X.Y., and Zhang, Z.H., Phosphorus, Sulfur Silicon Relat. Elem., 2008, vol. 183, p. 1672. https://doi.org/10.1080/10426500701724530
Das, B., Thirupathi, P., Reddy, K.R., Ravikanth, B., and Nagarapu, L., Catal. Commun., 2007, vol. 8, p. 535. https://doi.org/10.1016/j.catcom.2006.02.023
Das, B., Thirupathi, P., Mahender, I., Reddy, V.S., and Rao, Y.K., J. Mol. Catal. A: Chem., 2006, vol. 247, p. 233. https://doi.org/10.1016/j.molcata.2005.11.048
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares no conflict of interest.
Rights and permissions
About this article

Cite this article
Zeydi, M.M. Preparation of 1,3,5-Triazine-2,4,6-triaminium Trifluoromethanesulfonate and Its Use as an Eco-friendly Catalyst for the Synthesis of Xanthene Derivatives. Russ J Org Chem 58, 557–563 (2022). https://doi.org/10.1134/S1070428022040133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070428022040133