
Abstract
irst-generation dendrimers containing thiacalix[4]arene in cone and 1,3-alternate conformations as a core and hydroxyl groups as terminal moieties have been synthesized. It has been shown that self-assembly of the dendrimers with the macrocyclic core in cone conformation and terminal amidoethanol groups in an aqueous solution leads to the formation of stable monodisperse supramolecular system consisting of the nanoparticles with hydrodynamic diameter of 11.4±0.1 nm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Development of the systems for targeted drug delivery has remained among the essential directions at the edge between chemistry and medicine over recent decades [1–3]. Such systems allow overcoming the drawbacks of conventional therapeutic agents (small drug molecules, peptides, and genes) such as low solubility in aqueous media, high toxicity, poor stability, and poorly controllable or uneven release, which increase the therapeutic index of the drugs and decrease the undesirable side effects.
Nanosized targeted delivery systems are of special interest [4–6], since the systems with 30‒200-nm particles have been considered optimal for medical applications [7]. The dendrimers are advantageous among the nanosized drug delivery systems, due to their properties, e.g., multivalence, defined branched structure set at the synthesis stage, monodispersity, variable terminal groups, and the presence of internal voids between the dendrons, prone to binding of hydrophobic molecules [8, 9]. As far as drug delivery is concerned, dendrimers are flexible in the loading of the substrates: a drug can be noncovalently bound inside the macromolecule or at its surface or covalently linker to the dendrimer [10]. The aforementioned properties allow for targeted drug delivery by dendrimers.
The poly(amidoamine), poly(propylene imine), and poly(L-lysine) dendrimers are the most widely used water-soluble dendrimers. They contain terminal amino groups which can be easily modified to impart the desired properties to the macromolecule. A disadvantage of such dendrimers is significant increase in their cytotoxicity with the increase in the macromolecule generation number, due to the presence of large positive charge at the surface [11]. To resolve this issue, the dendrimer terminal groups can be modified with various fragments such as poly(ethylene glycol), carboxylic, hydroxyl, and acetyl ones [11]. Such modification has reduced the general toxicity of the macromolecule in comparison with the non-modified analogs containing terminal primary amino groups [12, 13]. However, the efficiency of the substrates binding with the modified dendrimers has been simultaneously decreased [12, 14]. Hence, the issue of preparation of low-toxic dendrimers efficiently binding the biological substrates has remained unresolved.
We have earlier suggested a method to synthesize first-generation poly(amidoamine) dendrimers based on thiacalix[4]arene [15] and their precursors, which is a convenient route to the branched macromolecules with predefined properties. However, these compounds have contained terminal primary amino groups, which can negatively affect their cytotoxicity. Therefore, to eliminate the above-marked disadvantages of the dendrimers and to ensure the low toxicity, we suggested the design of dendrimers containing a thiacalix[4]arene core and hydroxyl terminal groups. The presence of the nontoxic thiacalix[4]arene core, which can exist in three conformations (cone, partial cone, and 1,3-alternate) and thus ensure the presence of broad internal voids in the dendrimer, as well as large number of the binding sites (amide and terminal hydroxyl groups) should provide a dendrimer exhibiting low toxicity and efficiently binding various substrates.
Hence, in this study we suggested the synthesis of novel first-generation dendrimers based on the macrocyclic thiacalix[4]arene core in cone and 1,3-alternate conformations, containing terminal hydroxyl groups, and investigated their aggregation properties.
The previously obtained ester derivatives of thiacalixarene 1 and 2 in the cone and 1,3-alternate conformations, respectively, were used as the starting compounds for the synthesis of the target dendrimers bearing terminal hydroxyl groups. In order to introduce the hydroxyl groups, aminolysis of the compounds 1 and 2 with amino alcohol differing in the length of the alkylidene fragment (ethanolamine and propanolamine) was considered, to assess the influence of the spacer length on the aggregation ability. It is known that the synthesis of poly(amidoamine) dendrimers should be performed under mild conditions to minimize the side reactions such as retro-Michael reaction and intra- as well as intermolecular crosslinking via transesterification. Therefore, the aminolysis of the compounds 1 and 2 was carried out at temperature not exceeding 30°C (Scheme 1). The reaction was monitored by TLC. As a result, the first-generation dendrimers 3–6 containing ethylidene and propylidene spacers were isolated upon 80 h with yield of 50–69%. The reduced compounds yield in comparison with the dendrimers bearing terminal primary amino groups [15], was explained by the losses due to different isolation methods: since ethanolamine and propanolamine could not be efficiently removed due to their high boiling point, the products were isolated via re-precipitation in water, in which the target compounds were partially soluble.

1.
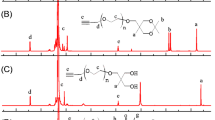
1Н NMR spectrum of the compound 3 in cone conformation contained the signals of the protons of the tert-butyl, oxymethylene, and aromatic fragments as singlets at 1.07, 4.75, and 7.38 ppm, respectively, whereas the methylene groups at the amide nitrogen atoms appeared as two multiplets at 3.04–3.12 and 3.12–3.20 ppm (Table 1). In the case of the compound 4 (1,3-alternate), the proton signals of the above-listed groups appeared at 1.19, 3.93, and 7.52 ppm, whereas the signals of the methylene groups at the amide nitrogen atoms were overlapped and appeared as a multiplet at 3.01– 3.12 ppm. Such shift of the signals is typical of the thiacalix[4]arene derivatives and is due to different mutual spatial location of the parts of the macromolecule and, hence, shielding of the groups sterically close to the macrocycle platform. The influence of the thiacalixarene platform was weakened with the increase in the substituent distance from it. For example, chemical shifts of the branches’ protons points as well as of the methylene groups at the tertiary nitrogen atom and the hydroxyl groups were not affected by the thiacalixarene conformation. It should be also noted that the signals of the protons of the branching methylene groups and the hexylidene fragments appeared as broadened multiplets in the 1Н NMR spectra of the obtained compounds 3–6. Therefore, the determination of the spin-spin interaction constants is difficult. Likely, the described spectral features were due to conformational flexibility of the dendrimer macromolecule and, overlap of the signals of the protons of different methylene groups. Overall, the values of chemical shifts of and the ratio of the integral intensities in the 1Н NMR spectra of the compounds 3–6 unambiguously confirmed the dendrimers structure.
IR spectra of the obtained dendrimers 3–6 contained a series of characteristic bands: the band at 1085 cm–1 was assigned to the vibrations of the aryl alkyl ether groups (CPhOCH2), the amide I and amide II bands at 1648 and 1538 cm–1 corresponded to the amide groups vibrations, and the broad bands at 3072 and 3290 cm–1 were due to the vibrations of the N–H and O–H bonds. Significant broadening of the absorption band at 3290 cm–1 likely reflected numerous intra- and intermolecular hydrogen bonds in the crystal structure.
It is interesting to note that the MALDI mass spectra of the obtained dendrimers 3–6 contained, besides the molecular ion peak, a sequential series of the signals corresponding to stepwise elimination of the dendrimer branches via the retro-Michael reaction occurring due to fragmentation of the molecular ion. For example, the mass spectrum of the compound 3 contained a weak peak at m/z 2290.765, assigned to the molecular fragment with a sodium ion [M + Na]+, and the peaks of the molecular ions with one (m/z 2175.438), two (m/z 2030.123), three (m/z 1944.815), four (m/z 1829.554), and five (m/z 1714.286) branches of the dendrimer eliminated (Scheme 2). Such pattern is typical of the MALDI mass spectra obtained during analysis of dendrimers [16, 17].

2.
At the next stage of the study, we investigated the aggregation ability of the obtained compounds 3–6 by dynamic light scattering (DLS) in a phthalate buffer solution (pH 4.01) at the dendrimer concentration ranging between 1×10–6 and 1×10–3 M. It is known that solubility of the derivatives of thiacalix[4]arene in water depends on the macrocycle conformation [18–20]. The solubility of the compound in the 1,3-alternate conformation is usually significantly poorer in comparison with the cone stereoisomers. Analogous trend was observed in this study: the compound 6 containing the propylidene spacers was insoluble under the considered conditions. It was found that the obtained compounds 3–5 at concentration of 1×10–6 to 1×10–4 M did not form stable colloidal systems, as evidenced by unsatisfactory correlation function values. At concentration 1×10–3 M, formation of stable supramolecular systems consisting of the nanosized particles was observed in the case of compounds the 3–5 (Table 2). The monodisperse systems with low polydispersity index (PDI) were formed only in the case of the dendrimer 3 containing the macrocyclic core in cone conformation: the mean hydrodynamic diameter of the particles equaled 11.4±0.1 nm (PDI 0.15). In the cases of the dendrimers 4 and 5, predominantly bimodal distribution was observed at concentration 1×10–3 M. The compound 5 (cone conformation) formed the nanoparticles with size of 7.5±0.2 nm as well as the particles with size of 393±81 nm, their ratio by intensity being 90 : 10 (PDI 0.32). In the case of the compound 4 with the macrocyclic core in 1,3-alternate conformation, mean sizes of the particles were 223±4 and 3487±1487 nm, their ratio by intensity being 82 : 18 (PDI 0.39).
The monomodal distribution observed in the case of the compound 3 could be explained by conformation of the macrocyclic core of the dendrimer (cone), in which the hydrophilic substituents of the lower rim of the macrocycle (the dendrimer branches) were to the same side of the macrocyclic platform, the hydrophobic substituents of the upper rim (the tert-butyl groups) being located to the other side. Such location of the substituents imparted amphiphilic properties to the obtained dendrimer, which likely led to the formation of the stable systems containing the nanoparticles with size of 11.4±0.1 nm and low polydispersity index. The dendrimer 5 was more lipophilic in comparison with the compound 3, due to elongation of the spacer between the amide and hydroxyl groups by a methylene group in each of eight terminal fragments, which resulted in the formation of the second fraction of the particles with size of 393± 81 nm, besides the nanoparticles with size of 7.5±0.2 nm, and, hence, increase in the system polydispersity. The compound 4 (1,3-alternate) contained the substituents distributed symmetrically with respect to the macrocyclic platform, and the macromolecule did not exhibit amphiphilic properties, which explained the formation of the systems containing larger aggregates, with sizes of 223.3±4.3 and 3487±1487 nm. It should be noted that the length of the spacer at the hydroxyl group significantly affected the aggregation properties of the obtained dendrimers. In particular, the extension of the spacer from the ethylidene to the propylidene in the case of the compounds 3 and 5 led to the increase in the system polydispersity, whereas in the case of the compounds 4 and 6 the same change resulted in significant decrease in the solubility.
In summary, novel first-generation dendrimers containing terminal hydroxyl groups with the thiacalix[4]arene core in cone and 1,3-alternate conformations were obtained. The formation of monodisperse systems containing nanoparticles (size 11.4±0.1 nm, PDI 0.15) was observed in an aqueous medium only in the case of the dendrimer with the macrocyclic core in cone conformation containing terminal amidoethanol fragments. It was shown that the increase in the spacer length in the obtained dendrimers by a methylene group led to significant decrease in the solubility in water. The obtained results can be used in the development of novel low-toxic systems based on stimulus-responsive nanoparticles for targeted drug delivery.
EXPERIMENTAL
1Н and 13С NMR spectra were recorded using a Bruker Avance 400 spectrometer operating at 400.0 and 100.0 MHz, respectively. The chemical shifts were determined relative to the signals of the residual protons of the deuterated solvent (DMSO-d6). Concentration of the analyzed solvents equaled 10 mM. The FTIR ATR spectra were recorded using a Spectrum 400 spectrometer (Perkin Elmer) with the following settings: resolution 1 cm–1, accumulation during 64 scans, registration during 16 s, and spectral range 400–4000 cm–1. The mass spectra were recorded using a MALDI-TOF Dynamo Finnigan mass spectrometer with p-nitroaniline as the matrix. The melting points were determined using a Boetius Block apparatus. Elemental analysis was performed using a PerkinElmer 2400 Series II instrument.
Size distribution of the particles formed during the first-generation dendrimers self-aggregation at 20°С was studied by dynamic light scattering using a Zetasizer Nano ZS analyzer (Malvern) in quartz cells. The instrument was equipped with a 4-mW He-Ne laser (wavelength 633 nm). The measurements were performed at the scattering angle 173°, the measuring position in the cell being determined automatically. The results were processed using DTS program (Dispersion Technology Software 4.20). The experiments were performed in a phthalate buffer solution (pH 4.1), the compounds concentration being varied between 1×10–6 and 1×10–3 M.
The starting thiacalix[4]arenes 1 and 2 containing eight ester groups at the lower rim were obtained as described elsewhere [15].
General procedure for the synthesis of the compounds 3 and 4. 0.21 mL (3.52 mmol) of ethanolamine was added to a solution of 0.22 g (0.11 mmol) of the starting compound 1 or 2 in 10 mL of methanol. The reaction mixture was stirred at 30°C during 80 h. The solvent was removed, and 20 mL of water was added to the residue. The formed precipitate was isolated by centrifugation and dried in vacuum over phosphorus pentoxide.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis[N-(6-{N,N-di[N-(2-hydroxyethyl)carbamoylethyl]amino}hexyl)carbamoylmethoxy]-2,8,14,20-tetrathiacalix[4]arene (3, cone). Yield 0.16 g (66%), mp 60°С. IR spectrum, ν, cm–1: 3289 (N–H, O–H), 3075 (N–H), 1644 [С(О)NH, amide I], 1541 [С(О)NH, amide II], 1094 (CPhOCH2). 1Н NMR spectrum, δ, ppm: 1.07 s [36H, (CH3)3C], 1.16–1.31 m [16H, C(O)NHCH2CH2CH2CH2, C(O)NHCH2CH2CH2CH2], 1.31–1.39 m (8H, CH2CH2N), 1.40–1.50 m [8H, C(O)NHCH2CH2CH2], 2.14–2.22 m [16H, NCH2CH2C(O)NH], 2.40–2.47 m (8H, CH2CH2N), 2.61–2.67 m [16H, NCH2CH2C(O)NH], 3.04–3.12 m (16H, NHCH2), 3.12–3.20 m (8H, NHCH2), 3.33–3.39 m (16H, CH2OH), 4.75 s [8H, OCH2C(O)], 7.38 s (8H, Ar-H), 7.95–8.01 m (8H, NH), 8.30–8.37 m (4H, NH). 13C NMR spectrum, δC, ppm: 26.57, 26.73, 29.22, 29.58, 30.77, 33.09, 33.93, 35.80, 38.53, 45.80, 49.17, 59.92, 73.81, 128.03, 134.41, 146.50, 157.77, 167.68, 171.51. Mass spectrum (MALDI), m/z: 2290.77 [M + Na]+. Found, %: C 59.04; H 8.61; N 10.19; S 5.35. C112H184N16O24S4. Calculated, %: C 59.34; H 8.18; N 9.89; S 5.66.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis[N-(6-{N,N-di[N-(2-hydroxyethyl)carbamoylethyl]amino}hexyl)carbamoylmethoxy]-2,8,14,20-tetrathiacalix[4]arene (4, 1,3-alternate). Yield 0.17 g (69%), mp 73°С. IR spectrum, ν, cm–1: 3290 (N–H, O–H), 3072 (N–H), 1648 [С(О)NH, amide I], 1538 [С(О)NH, amide II], 1085 (CPhOCH2). 1Н NMR spectrum, δ, ppm: 1.14–1.30 m [52H, (CH3)3C, C(O)NHCH2CH2CH2CH2, C(O)NHCH2CH2CH2CH2], 1.31–1.49 m [16H, CH2CH2N, C(O)NHCH2CH2CH2], 2.15–2.22 m [16H, NCH2CH2C(O)NH], 2.41–2.47 m (8H, CH2CH2N), 2.61–2.68 m [16H, NCH2CH2C(O)NH], 3.01–3.12 m (24H, NHCH2), 3.33–3.41 m (16H, CH2OH), 3.93 s [8H, OCH2C(O)], 7.52 s (8H, Ar-H), 7.65–7.72 m (4H, NH), 7.94–8.00 m (8H, NH). 13C NMR spectrum, δC, ppm: 26.12, 26.61, 29.17, 29.48, 30.78, 33.13, 33.89, 35.77, 38.80, 45.76, 49.10, 59.94, 70.40, 127.49, 132.08, 146.26, 156.53, 166.88, 171.59. Mass spectrum (MALDI), m/z: 2268.87 [M + H]+. Found, %: C 58.99; H 8.67; N 9.97; S 5.27. C112H184N16O24S4. Calculated, %: C 59.34; H 8.18; N 9.89; S 5.66.
General procedure for the synthesis of the compounds 5 and 6. 0.27 mL (3.52 mmol) of propanolamine was added to a solution of 0.22 g (0.11 mmol) of the starting compound 1 or 2 in 10 mL of methanol. The reaction mixture was stirred at 30°C during 80 h. The solvent was removed, and 20 mL of water was added to the residue. The formed precipitate was separated off on a Schott funnel and dried in vacuum over phosphorus pentoxide.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis[N-(6-{N,N-di[N-(2-hydroxypropyl)carbamoylethyl]amino}hexyl)carbamoylmethoxy]-2,8,14,20-tetrathiacalix[4]arene (5, cone). Yield 0.13 g (50%), mp 59°С. IR spectrum, ν, cm–1: 3289 (N–H, O–H), 3075 (N–H), 1648 [С(О)NH, amide I], 1544 [С(О)NH, amide II], 1094 (CPhOCH2). 1Н NMR spectrum, δ, ppm: 1.07 s [36H, (CH3)3C], 1.16–1.30 m [16H, C(O)NHCH2CH2CH2CH2, C(O)NHCH2CH2CH2CH2], 1.30–1.40 m (8H, CH2CH2N), 1.40–1.56 m [24H, C(O)NHCH2CH2CH2, NHCH2CH2CH2OH], 2.13–2.21 m [16H, NCH2CH2C(O)NH], 2.40–2.47 m (8H, CH2CH2N), 2.61–2.69 m [16H, NCH2CH2C(O)NH], 3.02–3.10 m (16H, NHCH2), 3.11–3.20 m (8H, NHCH2), 3.34–3.42 m (16H, CH2OH), 4.75 s [8H, OCH2C(O)], 7.38 s (8H, Ar-H), 7.92–7.98 m (8H, NH), 8.30–8.40 m (4H, NH). 13C NMR spectrum, δC, ppm: 26.57, 26.74, 29.23, 29.62, 30.77, 32.48, 33.93, 35.57, 35.87, 38.53, 45.88, 49.17, 58.40, 73.84, 128.04, 134.40, 146.50, 157.74, 167.67, 171.49. Mass spectrum (MALDI), m/z: 2418.0 [M + K]+. Found, %: C 60.24; H 8.83; N 9.87; S 5.21. C120H200N16O24S4. Calculated, %: C 60.58; H 8.47; N 9.42; S 5.39.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis[N-(6-{N,N-di[N-(2-hydroxypropyl)carbamoylethyl]amino}hexyl)carbamoylmethoxy]-2,8,14,20-tetrathiacalix[4]arene (6, 1,3-alternate). Yield 0.15 g (61%), mp 78°С. IR spectrum, ν, cm–1: 3300 (N–H, O–H), 3074 (N–H), 1648 [С(О)NH, amide I], 1538 [С(О)NH, amide II], 1086 (CPhOCH2). 1Н NMR spectrum, δ, ppm: 1.12–1.30 m [52H, (CH3)3C, C(O)NHCH2CH2CH2CH2, C(O)NHCH2CH2CH2CH2], 1.31–1.47 m [16H, CH2CH2N, C(O)NHCH2CH2CH2], 1.48–1.56 m (16H, NHCH2CH2CH2OH) 2.12–2.21 m [16H, NCH2CH2C(O)NH], 2.41–2.47 m (8H, CH2CH2N), 2.60–2.68 m [16H, NCH2CH2C(O)NH], 2.99–3.13 m (24H, NHCH2), 3.34–3.43 m (16H, CH2OH), 3.93 s [8H, OCH2C(O)], 7.52 s (8H, Ar-H), 7.64–7.73 m (4H, NH), 7.91–7.99 m (8H, NH). 13C NMR spectrum, δC, ppm: 26.60, 26.64, 29.17, 29.51, 30.78, 32.47, 33.90, 35.57, 35.82, 38.80, 45.82, 49.11, 58.40, 70.38, 127.48, 132.04, 146.26, 156.50, 166.88, 171.46. Mass spectrum (MALDI), m/z: 2417.9 [M + Na]+. Found, %: C 60.49; H 9.00; N 9.78; S 5.19. C120H200N16O24S4. Calculated, %: C 60.58; H 8.47; N 9.42; S 5.39.
REFERENCES
Tiwari, G., Tiwari, R., Bannerjee, S.K., Bhati, L., Pandey, S., Pandey, P., Sriwastawa, B., Int. J. Pharm. Investig., 2012, vol. 2, no. 1, p. 2. https://doi.org/10.4103/2230-973X.96920
Liu, D., Yang, F., Xiong, F., and Gu, N., Theranostics, 2016, vol. 6, no. 9, p. 1306. https://doi.org/10.7150/thno.14858
Sung, Y.K. and Kim, S.W., Biomater. Res., 2020, vol. 24, no. 1, p. 12. https://doi.org/10.1186/s40824-020-00190-7
Dang, Y. and Guan, J., Smart Mater. Med., 2020, vol. 1, p. 10. https://doi.org/10.1016/j.smaim.2020.04.001
Yao, Y., Zhou, Y., Liu, L., Xu, Y., Chen, Q., Wang, Y., Wu, S., Deng, Y., Zhang, J., and Shao, A., Front. Mol. Biosci., 2020, vol. 7, p. 193. https://doi.org/10.3389/fmolb.2020.00193
Amreddy, N., A. Babu, Muralidharan, R., Panneerselvam, J., Srivastava, A., Ahmed, R., Mehta, M., Munshi, A., and Ramesh, R., Adv. Cancer Res., 2018, p. 115. https://doi.org/10.1016/bs.acr.2017.11.003
Hao, M., Chen, B., Zhao, X., Zhao, N., and Xu, F.J., Mater. Chem. Front., 2020, vol. 4, no. 9, p. 2571. https://doi.org/10.1039/D0QM00323A
Castro, R.I., Forero-Doria, O., and Guzmán, L., An. Acad. Bras. Ciênc., 2018, vol. 90, no. 2, p. 2331. https://doi.org/10.1590/0001-3765201820170387
Mukherjee, S., Mukherjee, S., Abourehab, M.A.S., Sahebkar, A., and Kesharwani, P., Eur. Polym. J., 2022, vol. 177, p. 111471. https://doi.org/10.1016/j.eurpolymj.2022.111471
Kesharwani, P., Jain, K., and Jain, N.K., Prog. Polym. Sci., 2014, vol. 39, no. 2, p. 268. https://doi.org/10.1016/j.progpolymsci.2013.07.005
Hsu, H.J., Bugno, J., Lee, S., and Hong, S., WIREs Nanomed. Nanobiotechnol., 2016, vol. 9, no. 1, p. e1409. https://doi.org/10.1002/wnan.1409
Fant, K., Esbjörner, E.K., Jenkins, A., Grossel, M.C., Lincoln, P., and Nordén, B., Mol. Pharm., 2010, vol. 7, no. 5, p. 1734. https://doi.org/10.1021/mp1001312
Duncan, R. and Izzo, L., Adv. Drug Deliv. Rev., 2005, vol. 57, no. 15, p. 2215. https://doi.org/10.1016/j.addr.2005.09.019
Bodewein, L., Schmelter, F., Di Fiore, S., Hollert, H., Fischer, R., and Fenske, M., Toxicol. Appl. Pharmacol., 2016, vol. 305, p. 83. https://doi.org/10.1016/j.taap.2016.06.008
Mostovaya, O., Padnya, P., Shiabiev, I., Mukhametzyanov, T., and Stoikov, I., Int. J. Mol. Sci., 2021, vol. 22, no. 21, p. 11901. https://doi.org/10.3390/ijms222111901
Zhou, L., Russell, D.H., Zhao, M., and Crooks, R.M., Macromolecules, 2001, vol. 34, no. 11, p. 3567. https://doi.org/10.1021/ma001782j
Subbi, J., Aguraiuja, R., Tanner, R., Allikmaa, V., and Lopp, M., Eur. Polym. J., 2005, vol. 41, no. 11, p. 2552. https://doi.org/10.1016/j.eurpolymj.2005.05.031
Padnya, P.L., Andreyko, E.A., Gorbatova, P.A., Parfenov, V.V., Rizvanov, I.K., and Stoikov, I.I., RSC Adv., 2017, vol. 7, no. 3, p. 1671. https://doi.org/10.1039/C6RA24734B
Padnya, P.L., Terenteva, O.S., Akhmedov, A.A., Iksanova, A.G., Shtyrlin, N.V., Nikitina, E.V., Krylova, E.S., Shtyrlin, Y.G., and Stoikov, I.I., Bioorg. Med. Chem., 2021, vol. 29, p. 115905. https://doi.org/10.1016/j.bmc.2020.115905
Shurpik, D.N., Padnya, P.L., Stoikov, I.I., and Cragg, P.J., Molecules, 2020, vol. 25, no. 21, p. 5145. https://doi.org/10.3390/molecules25215145
Funding
This study was financially supported by the Russian Science Foundation (grant no. 21-73-20067). Registration of the mass spectra was supported by the Program for Strategic Academic Leadership of Kazan (Volga Region) Federal University (“Prioritet-2030”).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
To the 145th anniversary of A.E. Arbuzov
Supplementary information
Rights and permissions
About this article

Cite this article
Shiabiev, I.E., Pysin, D.A., Padnya, P.L. et al. First-Generation Dendrimers Based on Thiacalix[4]arene Containing Hydroxyl Terminal Groups: Synthesis and Self-Assembly. Russ J Gen Chem 92, 2574–2581 (2022). https://doi.org/10.1134/S1070363222120040
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363222120040