
Abstract
New iron(II) and copper(II) coordination compounds with 2,6-bis[1-(phenylimino)ethyl]pyridine (L1), Fe(L1)2SO4·H2O, Fe(L1)2(ClO4)2, Cu(L1)Cl2, and Cu(L1)2Br2·2H2O, were synthesized. The compounds were identified and investigated using CHN analysis and electronic spectroscopy (diffuse reflectance spectroscopy), IR spectroscopy, XRD, and static magnetic susceptibility. Antiferromagnetic exchange interactions between paramagnetic centers are observed in all iron and copper complexes in the ranges 80–420 and 1.77–300 K, respectively. Evaluation of the cytotoxic activity of copper(II) complexes showed that the Cu(L1)2Br2 complex (IC50 26.7 μmol/L) exhibits the highest activity against the human breast adenocarcinoma cell line (MCF-7).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Polynitrogen-containing heterocyclic compounds are a promising class of ligands for the synthesis of transition metal coordination compounds with magnetic, biological, and catalytic activity. Bisiminopyridine complexes are capable of catalyzing the ethylene polymerization to linear polyethylene [1–3], the reactions of [2+2] cycloaddition of unactivated olefins [4], the C‒H functionalization [5], the hydroboration and hydrosilylation of alkenes [6, 7], and the activation of small molecules (N2, O2, CO2) [8–10]. In bisiminopyridine complexes of metals with electronic configurations d4‒d7, a spin crossover (spin transition) can take place under specific conditions. The change in the spin multiplicity occurs under the influence of temperature, pressure, irradiation with light of a certain wavelength, and other factors. This class of complexes includes iron(II) compounds with nitrogen-containing ligands, in which the spin transition 1А1↔5Т2 is observed [11–13]. In Co(II), Ni(II), and Cu(II) polynuclear complexes with ligands of this class, antiferro- or ferromagnetic exchange interactions between paramagnetic centers are observed. A necessary condition for their occurring is cooperative interactions in the solid phase of the complexes [14]. The search for new molecular magnets is an important task of modern chemistry.
Potential nitrogen-containing ligands include pyridine derivatives—2,6-bis[1-(phenylimino)ethyl]pyridine (L1) and 2,6-bis(benzimidazol-2-yl)pyridine (L2, Scheme 1), which have the structure, which predetermines their tridentate cyclic mode of coordination to a central ion on complex formation. The coordination of two such ligands to a metal, in particular to Fe(II), leads to the formation of an octahedral polyhedron with a coordination node FeN6, which is a prerequisite for the spin crossover occurrence. We have previously synthesized iron(II) complexes [Fe(L2)2]Ai·nH2O (A is an anion; i = 1, 2; n = 0‒2), with the compound L2 and various anions in which the spin crossover 1А1↔5Т2 is observed [15–17]. It seemed ppropriate to continue studies with this class of ligands, in particular, with the compound L1. This ligand previously served as the basis for the synthesis of a number of compounds with double- and triple-charged 3d metal ions and Cd(II) [18–26]. Most of the synthesized complexes have the composition [M(L1)Ai] (i = 2, 3). The ligand is coordinated to the metal in a tridentate-cyclic type by three nitrogen atoms, the coordination nodes are supplemented up to five by halide ions (Cl–, Br–) or oxygen atoms of the nitrate ion [26, 27]. Complexes with two L1 ligands, [Ni(L1)2](BF4)2 [26] and [Cu(L1)2](ClO4)2 [27], were obtained. Most complexes with the L1 ligand exhibit catalytic and luminescent properties; their magnetic and biological activity has not been studied.

1.
We synthesized Fe(II) and Cu(II) complexes with the ligand L1, Fe(L1)2SO4·H2O, Fe(L1)2(ClO4)2, Cu(L1)Cl2, and Cu(L1)2Br2·2H2O, and investigated their magnetic and cytotoxic properties. Complexes Fe(L1)2SO4·H2O (1), Fe(L1)2(ClO4)2 (2), and Cu(L1)2Br2·2H2O (4) were isolated from aqueous-organic solutions at the stoichiometric ratio M : L1 = 1 : 2. For the synthesis of complex [Cu(L1)Cl2] (3) under the same conditions, the ratio M : L1 = 1:2 was also used, however, a complex with the ratio M : L1 = 1 : 1 was obtained. When synthesizing Fe(II) complexes, to preserve iron in the lowest oxidation state, ascorbic acid was added to solutions as a reducing agent and a weak acidifying reagent. The compound [Fe(L1)2](ClO4)2 was synthesized in two stages. At the first stage, a Fe(ClO4)2 solution was obtained by merging aqueous solutions of FeSO4 and Ba(ClO4)2; at the second stage, a reaction between the Fe(ClO4)2 and L1 solutions occurred. The resulting complexes are stable when stored in air and decompose without melting when heated to 450 K.
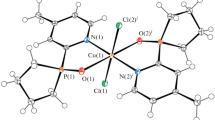
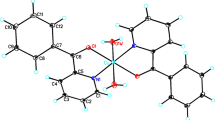
The Cu(L1)Cl2 complex was obtained earlier [26]. According to X-ray diffraction data, ligand L1 in this complex is coordinated to Cu(II) by three nitrogen atoms, the structure of the coordination polyhedron is complemented to bipyramidal by two chloride ions to form the CuN3Cl2 node. The same way of L1 coordination was found in the above-mentioned complexes with two L1 ligands: [Cu(L1)2](ClO4)2 [26] and [Ni(L1)2](BF4)2 [27]. These compounds have a distorted octahedral structure of the coordination polyhedron with the MN6 node; the anions occupy the outer-sphere position.
The main vibrational frequencies in the IR spectra of the compound L1 and complexes are given in Table 1. In the range 3200–3050 cm–1 there are stretching vibrations of NH groups, in the range 3100–2850 cm–1—stretching vibrations ν(CH) and ν(CH3), and in the range 1650– 1450 cm–1—stretching and bending vibrations of benzene and pyridine cycles. In the spectra of the complexes in the range of ring vibrations, the number and position of bands differ from those in the spectrum of compound L1, which points to the coordination of nitrogen atoms of pyridine and imino groups to the metal ions. This conclusion is confirmed by the spectra in the far-region, where the bands of metal–ligand vibrations appear, which are absent in the compound L1 spectrum. This range contains bands that can be attributed to the ν(M–N) and ν(M–Cl) vibrations (Table 1).
The magnetic susceptibility χp(T) of the CuLCl2 complex points to its paramagnetism and in the temperature range 50–300 K can be formally described by the Curie–Weiss dependence with the effective moment µeff ≈ 1.83 µB and θ ≈ ‒5 K (Fig. 1). However, when the interval of data processing is shifted towards low temperatures, the value of θ decreases by a factor of more than 20, and the dependence χp(T) shows no signs of antiferromagnetic ordering up to 1.77 K. This indicates that the real antiferromagnetic exchange interaction between copper ions is much weaker than would be expected from the value of θ obtained at high temperatures. The picture is clarified by the temperature dependence of the effective moment µeff calculated for θ equal to zero (Fig. 1). The value of µeff at 300 K is close to the theoretical value of 1.73 µB for spin-only (S = 1/2) moments of Cu2+ ions; the slight excess is due to the contribution of the orbital angular moments. As the temperature decreases to ~ 10 K, µeff smoothly decreases, which may point to the “freezing out” of orbital angular moments with µeff tending to the spin-only value of 1.73 µB. A sharp decrease in µeff at the lowest temperatures (below 10 K) points to a weak antiferromagnetic interaction between Cu2+ ions.

(a) Temperature dependence of the magnetic susceptibility χ for the CuLCl2 complex measured in the magnetic field H of (1) 1 and (2) 10 kOe, (b) the temperature dependence of the paramagnetic part of the susceptibility (in coordinates 1/χp) and the temperature dependence of the effective magnetic moment µeff calculated in the approximation of non-interacting moments (θ = 0).
For the CuL2Br2·2H2O complex, the magnetic susceptibility χp(T) in the temperature range 20–300 K is well described by the Curie–Weiss law, µeff ≈ 2.24 µB and θ ≈ ‒1 K (Fig. 2). The resulting effective moment significantly exceeds the theoretical value of 1.73 µB for spin-only (S = 1/2) moments of Cu2+ ions and points to a large contribution of orbital moments. The decrease in µeff observed at the lowest temperatures (<10 K) points to a weak antiferromagnetic interaction of the copper moments.

(a) Temperature dependence of the magnetic susceptibility χ for the CuL2Br2·2H2O complex measured in the magnetic field H of (1) 1 and (2) 10 kOe, (b) the temperature dependence of the paramagnetic part of the susceptibility (in coordinates 1/χp) and the temperature dependence of the effective magnetic moment µeff calculated in the approximation of non-interacting moments (θ = 0).
The temperature dependences of the magnetic susceptibility of the Fe(L1)2SO4∙H2O complex were studied in the range of 80-400 K (Figs. 3 and 4). At ~420 K, the compound decomposes. Spin crossover is not observed regardless of the presence or absence of crystallization water in the compound.

(a) Temperature dependence of the magnetic susceptibility χ for the Fe(L1)2SO4 complex (recording in an open ampoule); (b) 1/χ and µeff calculated in the noninteracting spin approximation.

(a) Temperature dependence of the magnetic susceptibility χ for the [Fe(L1)2]SO4.H2O complex (recording in a sealed ampoule); (b) 1/χ and µeff calculated in the noninteracting spin approximation.
The negative sign of θ for the dehydrated complex (‒25 ± 3 K) and for the crystallohydrate (‒47 ± 3 K) points to the antiferromagnetic interaction between iron magnetic moments in the substance. The values of the effective magnetic moment calculated in the noninteracting spin approximation (θ 0, Figs. 3 and 4) lie in the ranges of ~2.5‒2.7 and ~2.1‒2.4 µB. The values corresponding to these ranges, µeff 2.87 ± 0.03 µB for [FeL2]SO4 and µeff 2.59±0.03 µB for [FeL2]SO4∙H2O obtained with regard to θ, are less than the theoretical “spin-only” value of 4.9 µB for Fe(II). If we assume that the compound contains iron(II) ions both in the high-spin (HS) and low-spin (LS) states, then based on the µeff values obtained with regard to θ, we can estimate the ratio HS : LS = 0.34 : 0.66 for dehydrated complex and HS : LS = 0.28 : 0.72 for the crystallohydrate. Thus, dehydration of the complex is accompanied by an increase in µeff and a weakening of antiferromagnetic interactions.
Negative values of the static magnetic susceptibility of the Fe(L1)2(ClO4)2 complex are observed in the temperature range up to 420 K. With a further increase in temperature, intense decomposition of the compound begins. Thus, the Fe(II) ions in the studied compound are in the low-spin state, µeff 0, and the compound itself is diamagnetic both in the presence of water of crystallization and in the dehydrated state (Fig. 5).

Temperature dependences of the magnetic susceptibility χ for the Fe(L1)2(ClO4)2 complex. Recording in (a) an open ampoule and (b) a sealed ampoule.
The XRD data showed that all the obtained complexes are crystalline; however we failed to grow their single crystals. Based on indirect methods and comparison with published data, it can be concluded that compound L1 in the synthesized complexes with two ligands is coordinated in the same way as in the [Cu(L1)2](ClO4)2 [26] and [Ni(L1)2](BF4)2 [27] complexes with the formation of a distorted octahedral coordination polyhedron and the node MN6 (M = Fe, Cu). This is confirmed by the character of the diffuse reflectance spectra. In the diffuse reflectance spectra of Fe(L1)2SO4∙H2O, the bands at 485 and 745 nm can be attributed to d‒d transitions 1A1→1T1 (20619 cm–1) and 5T2→5E (13423 cm–1) in distorted octahedral iron(II) complexes with the coordination node FeN6 [28]. Consequently, the complex contains both the low-spin 1A1 form and the high-spin 5T2 form in the ratio calculated from the magnetochemical data. In the diffuse reflectance spectra of Fe(L1)2(ClO4)2, the only band at 744 nm corresponds to the d‒d transition 5T2→5E (13440 cm–1), which points to the high-spin state of the complex.
The effect of copper(II) complexes on the viability of HepG2 and MCF-7 human cells was assessed in the presence of the test compounds dissolved in ethanol by the method of double staining with Hoechst 33342/PI followed by differentiation of cells into living, dead, and apoptotic. Semi-maximal inhibition (IC50), i.e. the concentration of the drug at which cell death is 50%, was calculated after nonlinear approximation of the curves of experimental dependence of cell survival on the substance concentration.
Copper(II) complexes showed no cytotoxic activity against the HepG2 cell line (Fig. 6), but exerted a cytostatic effect. The action of the [CuL1Cl2] complex, starting from its concentration of 25 µmol/L, led to a slight decrease in the rate of cell growth, whereas the action of the 50 μmol/L [Cu(L1)2]Br2·2H2O solution twice reduced it in comparison with a control. The MCF-7 cell line was found to be more sensitive to the effects of the complexes under study (Fig. 7). The action of a 25 μmol/L solution of copper(II) bromide with L1 on the MCF-7 cell line caused the death of ~50% of cells (IC50 26.7 ± 0.5 μmol/L), whereas the maximal experimental concentration of copper (II) chloride with L1 caused the death of ~10% of cells. Thus, in relation to the both cell lines, copper(II) bromide had a more active effect on cell viability as compared to copper(II) chloride.

Influence of complexes of copper(II) halides with 2,6-bis[1-(phenylimino) ethyl]pyridine on the viability of HepG2 cells. (1) Amount of cells, (2) dead cells, (3) living cells, and (4) apoptosis.

Influence of complexes of copper(II) halides with 2,6-bis [1-(phenylimino)ethyl]pyridine on the viability of MCF-7 cells. (1) Amount of cells, (2) dead cells, (3) living cells, and (4) apoptosis.
EXPERIMENTAL
Commercial metal salts and solvents were used in the synthesis without further purification.
The IR absorption spectra of the complexes were recorded on Scimitar FTS 2000 and Vertex 80 IR Fourier spectrometers in the ranges 4000–400 cm–1 and 400–100 cm–1, respectively. The samples were prepared in the form of suspensions in Vaseline and fluorinated oils and in polyethylene. The diffuse reflectance spectra were recorded on a UV-3101 PC Shimadzu scanning spectrophotometer at room temperature in the range 300–2000 nm. The 1H NMR spectra were recorded on a Bruker AV-400 instrument with an operating frequency of 400.13 MHz.
The static magnetic susceptibility of copper complexes was measured on a Quantum Design MPMS-XL SQUID magnetometer in the temperature range 1.77–300 K and magnetic field H range 0–10 kOe. Magnetic properties of the iron complexes were studied by the Faraday method in the temperature range 80–420 K at H 0–7.3 kOe. To study the dehydrated iron complexes, the samples were placed in open quartz ampoules and evacuated to a residual pressure of 10–2 mmHg in the measuring cell of the installation, then an inert atmosphere of helium at a pressure of 5 mmHg was created. When studying iron complexes containing crystallization water, the samples were sealed in quartz ampoules with atmospheric air.
The temperature-independent contribution χd was calculated using the Pascal additive scheme. To determine the effective magnetic moment of copper and iron ions µeff and the Weiss constant θ, the temperature dependences of the paramagnetic contribution to the magnetic susceptibility χp(T) = χ(T)–χd were analyzed using the Curie‒Weiss dependence (1).
Here NA and kB are Avogadro’s number and Boltzmann’s constant, respectively (µeff =[(3kB/NA)χp(Т – θ)]1/2). The values of the Weiss constant θ obtained in the processing made it possible to estimate the exchange interaction parameters.
Biological research was performed on human cell lines HepG2 (hepatocellular carcinoma) and MCF-7 (breast adenocarcinoma). Cell viability was evaluated by the method of double staining with Hoechst 33342/propidium iodide (PI) according to the standard method described earlier [29]. The cells were seeded on 96-well plates and cultured in the IMDM medium (Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (HyClone, Germany) in a CO2 incubator at 37°C. After 24 h, preparations dissolved in EtOH were added in the concentration range of 1–50 μmol/L and incubated for 48 h. The cells were stained with Hoechst 33342 fluorescent dyes (Sigma-Aldrich) and propidium iodide (Invitrogen) for 30 min at 37°C. The survey was performed on an IN Cell Analyzer 2200 (GE Healthcare, UK) in an automatic mode, 4 fields per well. Images were processed using In Cell Investigator software to determine living (normal nuclei—blue non-condensed chromatin evenly distributed throughout the nucleus), dead (red, enlarged nuclei with smooth normal structure or bright red with slightly condensed chromatin), and apoptotic (round cells, bright blue strongly condensed or fragmented chromatin) cells in the entire population. The result is presented as the percentage of cells from three independent experiments ± standard deviation.
2,6-Bis[1-(phenylimino)ethyl]pyridine (L1) was synthesized with 70% yield by refluxing 2,6-diacetylpyridine with an excess of aniline in methanol in the presence of catalytic amounts of formic acid as described in [30]. 1Н NMR spectrum (400 MHz, CDCl3), δ, ppm: 2.40 s (6H, Me), 6.84 d (4HAr, J 7.9 Hz), 7.11 t (2HAr, J 7.3 Hz), 7.37 t (4HAr, J 7.7 Hz), 7.86 t (1H, H4Py, J 7.8 Hz), 8.34 d (2H, H3.5Py, J 7.8 Hz).
[Fe(L1)2]SO4·H2O (1). A weighed portion of 1 mmol (0.28 g) of FeSO4·7H2O with addition of 0.1 g of ascorbic acid was dissolved with heating in 10 mL of water, and 2 mmol (0.62 g) of L1 was dissolved in 10 mL of methylene chloride, the solutions were heated and mixed. The resulting red solution was evaporated until a precipitate began to form. After cooling the solution with the precipitate in a crystallizer with ice, the red-violet precipitate was filtered off, washed two times with small portions of water, and dried in air. Yield 20%. Found, %: C 63.5; H 5.6; N 9.2. C42H40FeN6O5S. Calculated,%: C 63.3; H 5.1; N 10.5.
[Fe(L1)2](ClO4)2 (2). Weighed portions of 1 mmol (0.28 g) of FeSO4·7H2O and 1 mmol (0.34 g) of Ba(ClO4)2 with addition of 0.1 g of ascorbic acid were dissolved separately with heating in 10 mL of water each, then the resulting solutions were mixed. The BaSO4 precipitate was filtered off, and a solution of 2 mmol (0.62 g) of L1 in 10 mL of methylene chloride was added to the resulting solution of Fe(ClO4)2. The resulting red solution was evaporated until a precipitate began to form. After cooling the solution with the precipitate in a crystallizer with ice, the red-violet precipitate was filtered off, washed two times with small portions of methylene chloride, and dried in air. Yield 11%. Found, %: C 57.7; H 4.4; N 9.4. C42H38Cl2FeN6O8. Calculated, %: C 57.2; H 4.3; N 9.5.
[Cu(L1)Cl2] (3). A weighed portion of 1.5 mmol (0.27 g) of CuCl2 was dissolved with heating in 5 mL of ethanol, and 3 mmol (0.93 g) of compound L1—in 15 mL of methylene chloride; the solutions were mixed. To the resulting green solution, 10 mL of hexane was added. On cooling the solution, a green precipitate of the complex formed, which was washed two times with small portions of ethanol, and dried in air. Yield 55%. Found, %: C 55.9; H 4.3; N 9.2. C21H19Cl2CuN3. Calculated, %: C 56.3; H 4.3; N 9.4.
[Cu(L1)2]Br2·2H2O (4). Weighed portions of 0.5 mmol (0.11 g) of CuBr2 and 1 mmol (0.31 g) of compound L1 were dissolved with heating in 5 mL of ethanol and 10 mL of methylene chloride, respectively, and then the resulting solutions were mixed. The resulting brown solution was boiled to remove excess solvent. After cooling the solution in a crystallizer with ice, a brown precipitate formed, which was filtered off, washed two times with small portions of ethanol, and dried in air. Yield 48%. Found, %: C 56.5; H 4.5; N 9.6. C42H42Br2CuN6O2. Calculated, %: C 56.9; H 4.8; N 9.5.
REFERENCES
Flisak, Z. and Sun, W.-H., ACS Catal., 2015, vol. 5, p. 4713. https://doi.org/10.1021/acscatal.5b00820
Gibson, V.C., Redshaw, C., and Solan, G.A., Chem. Rev., 2007, vol. 107, p. 1745. https://doi.org/10.1021/cr068437y
Oleinik, I.I., Oleinik, I.V., Abdrakhmanov, I.B., Ivanchev, S.S., and Tolstikov, G.A., Russ. J. Gen. Chem., 2004, vol. 74, no. 10, p. 1575. https://doi.org/10.1007/s11176-005-0059-7
Bouwkamp, M.W., Bowman, A.C., Lobkovsky, E., and Chirik, P.J., J. Am. Chem. Soc., 2006, vol. 128, p. 13340. https://doi.org/10.1021/ja064711u
Tondreau, A.M., Stieber, S.C.E., Milsmann, C., Lobkovsky, E., Weyhermüller, T., Semproni, S.P., and Chirik, P.J., Inorg. Chem., 2013, vol. 52, p. 635. https://doi.org/10.1021/ic301675t
Obligacion, J.V. and Chirik, P.J., J. Am. Chem. Soc., 2013, vol. 135, p. 19107. https://doi.org/10.1021/ja4108148
Hojilla Atienza, C.C., Tondreau, A.M., Weller, K.J., Lewis, K.M., Cruse, R.W., Nye, S.A., Boyer, J.L., Delis, J.G.P., and Chirik, P.J., ACS Catal., 2012, vol. 2, p. 2169. https://doi.org/10.1021/cs300584b
Margulieux, G.W., Turner, Z.R., and Chirik, P.J., Angew. Chem. Int. Ed., 2014, vol. 53, p. 14211. https://doi.org/10.1002/anie.201408725
Badiei, Y.M., Siegler, M.A., and Goldberg, D.P., J. Am. Chem. Soc., 2011, vol. 133, p. 1274. https://doi.org/10.1021/ja109923a
Rummelt, S.M., Zhong, H., Korobkov, I., and Chirik, P.J., J. Am. Chem. Soc., 2018, vol. 140, p. 11589. https://doi.org/10.1021/jacs.8b07558
Spin Crossover in Transition Metal Compounds, vols. I–III, Gutlich, P. and Goodwin, H., Eds., Berlin: Springer, 2004, p. 233.
Halcrow, M.A., Spin-Crossover Materials Properties and Applications, Chichester: John Wiley & Sons Ltd, 2013.
Shakirova, O.G. and Lavrenova, L.G., Crystals, 2020, vol. 10, p. 843. https://doi.org/10.3390/cryst10090843
Buchachenko, A.L., Russ. Chem. Bull., 2011, vol. 60, no. 12, p. 2439. https://doi.org/10.1007/s11172-011-0378-2
Lavrenova, L.G., Dyukova, I.I., Korotaev, E.V., Sheludyakova, L.A., and Varnek, V.A., Russ. J. Inorg. Chem., 2020, vol. 65, no. 1, p. 30. https://doi.org/10.1134/S0044457X20010109
Ivanova, A.D., Korotaev, E.V., Komarov, V.Yu., Sheludyakova, L.A., Varnek, V.A., and Lavrenova, L.G., New J. Chem., 2020, vol. 44, p. 5834. https://doi.org/10.1039/d0nj00474
Ivanova, A.D., Lavrenova, L.G., Korotaev, E.V., Trubina, S.V., Sheludyakova, L.A., Petrov, S.A., Zhizhin, K.Yu., and Kuznetsov, N.T., Russ. J. Inorg. Chem., 2020, vol. 65, no. 11, p. 1687. https://doi.org/10.31857/S0044457X20110070
Restivo, R.J. and Ferguson, G., Dalton Trans., 1976, p. 518.
Bluhm, M.E., Folli, C., and Döring, M., J. Mol. Cat. A, 2004, vol. 212, p. 13. https://doi.org/10.1016/j.molcata.2003.11.003
Fan, R-Q., Zhu, D-S., Mu, Y., Guang-Hua, Li, G.-H., Yang, Y-L., Su, Q., and Feng, S.-H., Eur. J. Inorg. Chem., 2004, no. 24, p. 2304. https://doi.org/10.1002/ejic.200400443
Gong, D., Baolin, Wang, B., Bai, C., Bi, J., Wang, F., Dong, W., Zhang, X., and Jiang, L., Polymer, 2009, vol. 50, p. 6259. https://doi.org/10.1016/j.polymer.2009/10.054
Adnan, Abu-Surrah, Khalid, A.I., Maher, Y.A., and Ayman, A.I., J. Polym. Res., 2011, vol. 18, p. 59. https://doi.org/10.1007/s10965-010-9391-7
Edwards, D.A., Mahon, M.F., Martin, W.R., Molloy, K.C., Fanwick, P.E., and Walton, R.A., J. Chem. Soc. Dalton Trans., 1990, p. 3161.
Guo, M.-P., Gui-Quan Guo, G.-Q., Guo, H.-R., and Di-Chang Zhong, D.-C., Acta Crystallogr. E, 2007, vol. 63, p. m3025. https://doi.org/10.1107/S1600536807056644
Fan, R.-Q., Fan, R.-J., Lv, Z.-W., Lin Yang, Y.-L., An, F., and Gu, D.-M., J. Coord. Chem., 2007, vol. 60, no. 9, p. 919. https://doi.org/10.1080/00958970600914267
Trivedi, M., Pandey, D.S., and Xu, Q., Inorg. Chim. Acta, 2007, vol. 360, p. 2492. https://doi.org/10.1016/j.ica.2006.12.031
Blake, A.J., Lavery, A.J., Schröder, M., Acta Crystallogr. C, 1996, vol. 52, p. 37. https://doi.org/10.1107/S0108270195009851
Lever, A.B.P., Inorganic Electronic Spectroscopy, Amsterdam: Elsevier, 1985.
Solovieva, A.O., Vorotnikov, Yu.A., Trifonova, K.E., Efremova, O.A., Krasilnikova, A.A., Brylev, K.A., Vorontsova, E.V., Avrorov, P.A., Shestopalova, L.V., Poveshchenko, A.F., Mironov, Yu.V., and Shestopalov, M.A., J. Mater. Chem. B, 2016, vol. 4, p. 4839. https://doi.org/10.1039/C6TB00723F
Guo, J., Wang, B., Bi, J., Zhang, C., Zhang, H., Bai, C., Hu, Y., and Zhang, X., Polymer, 2015, vol. 59, p. 124. https://doi.org/10.1016/j.polymer.2015.01.006
Funding
This work was supported by the Russian science foundation (grant no. 20-63-46026) and the Ministry of science and higher education of the Russian Federation (projects nos. 121031700313-8, 121031700314-5, and 121011490013-7). Experiments on the analysis of cytotoxicity were carried out using the equipment of the Center for collective use “Proteomic analysis” with the support of the Ministry of education and science of Russia (agreement no. 075-15-2021-691).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Additional information
Translated from Zhurnal Obshchei Khimii, 2021, Vol. 91, No. 11, pp. 1693–1703 https://doi.org/10.31857/S0044460X21110068.
Rights and permissions
About this article

Cite this article
Lavrenova, L.G., Mishchenko, A.A., Oleynik, I.V. et al. Synthesis and Properties of Iron(II) and Copper(II) Coordination Compounds with 2,6-Bis[1-(phenylimino)ethyl]pyridine. Russ J Gen Chem 91, 2167–2175 (2021). https://doi.org/10.1134/S1070363221110062
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363221110062