
Abstract
Kinetic regularities and the mechanism of thermal decomposition of 3-substituted 1-(dinitromethyl)1H-1,2,4-triazoles in the solid phase and in dibutyl phthalate solution have been elucidated. The activation parameters have been determined, and the correlation dependencies between the logarithm of the rate constant of the limiting stage and the Hammett constant of substituents as well as the acid pKa value have been revealed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The interest to the study of kinetics of thermal decomposition of 3-substituted 1-(dinitromethyl)-1H-1,2,4-triazoles, relatively strong organic acids, is related to the synthesis of diverse energy-rich compounds and the investigation of the substation effect on the azoles reactivity towards thermal decomposition which is demanded for their safe use, storage, and targeted synthesis.
Herein we investigated kinetics of thermal decomposition of compounds 1‒6 (Scheme 1) to elucidate the influence of the nature of the substituent R on the rate, mechanism, and activation parameters of the rate-limiting stage.

1.
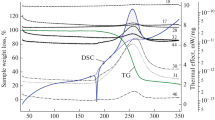
Using compounds 1 and 6 as examples, we performed differential thermogravimetric analysis to elucidate the influence of the substance phase state on the character and features and rate of the heat-induced decomposition as well as the temperature of the highest decomposition rate. Analysis of the thermograms revealed that thermal decomposition of compounds 1 and 6 occurred well before their melting points (at 82 and 78°С, respectively) and exhibited strong exothermic effect (peak temperature 86°С for compound 1 and 82°С for compound 6) as well as significant mass loss (by 50‒55%). That stage was followed by smooth mass decrease to 5‒7% of the starting value (Fig. 1). Sharp exothermic mass loss at the first stage led to degeneration of the endothermic melting peaks at 107°С (1) and 116°С (6). Such thermograms are typical of the autocatalytic thermal decomposition.

Thermogram of compound 6: (1) TG and (2) DTA.
Thermal decomposition of compound 6 under isothermal conditions below the melting point occurred with acceleration (Fig. 2, 1). The decomposition rate constant depended on the m/V ratio (m being the specimen mass, g, and V being the reaction vessel volume, cm3). In the case of compound 6 at 90°С, the increase in the m/V ratio by 50 times led to the increase in the rate constant by 5 times, and the induction period was shortened by 6.5 times. The dependence of the rate constant and the induction period on the m/V ratio was likely due to the autocatalytic thermal decomposition of the compound.

Thermal decomposition of compound 6 at 100°С: (1) in solid phase and (2) in 2 wt % solution in dibutyl phthalate.
To confirm that suggestion and estimate the true reactivity, we investigated thermal decomposition of compounds 1‒6 in an inert heat-resistant low-polar solvent (dibutyl phthalate, ε = 6.44), the use of which eliminated the influence of the crystal lattice and the interaction of the decomposition products with the starting compounds. That experiment showed that the decomposition features in the solution and in the solid phase were similar, yet the final gas volume (V∞, cm3/g) was decreased about twofold (Fig. 2, 2).
Chromatographic analysis of the solid residue formed upon decomposition of compounds 3 and 6 (conversion 93‒97%) revealed the presence of 3-chloro- and 3-nitro-1Н-1,2,4-triazole, respectively, in the case of the decomposition in a 2 wt % solution in dibutyl phthalate and the traces of those compounds in the case of decomposition of compound 3 in melt and compound 6 in the solid phase. That fact explained the difference in the gases evolution during thermal decomposition in the solution and in the solid phase (Fig. 2). Major gaseous products of the thermal decomposition of compounds 3 and 6 in the solution and in the solid phase were NO, N2O, and CO2.
The decrease in the compound concentration in the solution from 20 to 2 wt % led to the decrease in the thermal decomposition rate constant. Further dilution marginally affected the rate constant. That fact pointed at the existence of ionic pairs or larger ionic aggregates in the solution in equilibrium with the solvated ions rather than individual molecules. In view of that, the effect of temperature on the decomposition rate was investigated using 2 wt % solutions in dibutyl phthalate (Fig. 3).

Kinetics of thermal decomposition of compound 6 in a solution in dibutyl phthalate. (1) 100, (2) 110, (3) 120, and (4) 130°С.
The determined kinetic parameters, the Hammett constants σp, and ionization constants of certain CH-acids (the latter were taken from [1]) are collected in Table 1. Analysis of the collected data revealed a characteristic feature: the ksolution/ksolid ratio was 1– 2 orders of magnitude below unity. It should be noted that the ksolution/ksolid ratio for various high-energy compounds is generally above unity [2, 3], due to retarding effect of the crystal lattice [4].
Thermal decomposition of organic nitro compounds in the solid phase occurs via the dislocation mechanism at the boundaries of microblocks (size not exceeding 100 nm), and the ksolution/ksolid are of 5‒50 [5]. The increase in the crystal defectiveness likely observed in this study could be due to destruction of the crystals joints during heating as well as due to capture of moisture or solvent during purification via recrystallization [6]. Another possible reason for the increase in the dislocations number during thermal decomposition was the crystal etching by moisty compounds 1 and 6 (strong acids, Table 1).
The data in Table 1 showed that the increase in the substituent σp constant led to the increase in the thermal decomposition rate constant. The data points were linearized in the log k120°C‒σp coordinates following the regression Eq. (1).
The рKа value of the СН-acids is linearly dependent on the Hammett constant of the substituent σp [1]. In that case, the logarithm of the decomposition rate constant should be linear with рKа; that was indeed observed for the collected data, see regression Eq. (2). The compensation kinetic effect expressed by Eq. (3) was revealed for the studied СН-acids 1‒6.
Thermal decomposition of compounds 1‒6 was characterized by low activation parameters (Table 1), significantly different from those for 1-(R-dinitromethyl)-3-nitro-1H-1,2,4-triazoles [7], decomposition of which occurred via the homolytic mechanism with rupture of the C‒NO2 bond in the gem-dinitromethyl group. The earlier mentioned observations (weak influence of the solution concentration and experiment temperature on the rate constant, see Table 1) as well as negative activation entropy and the rate constant dependence on the рKа for compounds 1‒6 suggested that the heterolytic mechanism of the thermal decomposition was more likely. Therefore, we followed the example of 5-(dinitromethyl)-2-methyl-2Н-1,2,3-triazole [8], thermal decomposition of which in low-polar aprotic dibutyl phthalate occurred via the auto protolytic mechanism and was limited by the basic catalysis reaction (4), and expressed the thermal decomposition of compounds 1‒6 with the reactions series (4)‒(8) (Scheme 2). Further transformations (9)‒ (10) (Scheme 3) of radicals A and C corresponded to general view on the reaction of free radicals with NO and NO2.

2.

3.
Mechanism of auto protolytic thermal decomposition of compounds 1‒6 below the melting point could hardly be elucidated in full detail. However, it was clear that the CH-acids ionization occurred in the liquid water which was either formed during the decomposition or was present in the initial substance as occluded or crystal hydrate water [9]. The presence of water in the solid crystal led to destruction of the molecular crystal lattice at elevated temperature (80‒100°С) [9] and at the same time to relatively fast ionization [reaction (4)]. The high temperature of the experiment favored the ion-radical reactions [Eqs. (5) and (6)] leading to water regeneration. Further free radical reactions (9) and (10) gave the unstable 3-R-1,2,4-triazole radical [reaction (12)], which afforded hydrogen cyanide and the solid residue via the azole ring opening [reaction (13)]. The appearance of 1 mol of hydrogen cyanide (as confirmed by means of IR spectroscopy) explained the difference in the overall gas formation (V∞, Fig. 2) in the cases of the solid phase and the solution. In the latter case, the 3-R-1,2,4-triazole radical was stabilized by the transformation into 3-R-1Н-1,2,4-triazole via reaction (14). Since 3-R-1Н-1,2,4-triazoles were weaker acids (рKа ≥ 6.05 [10]) than compounds 1‒6, they acted as bases B [catalysts of the acid-base reaction (4)]. Ionization of compounds 1‒6 in the solution was 20–25 slower in comparison with the solid phase (where water vapor was accumulated at the dislocations) [11], and the decomposition was accelerated due to the appearance of water [reaction (8)] and 3-R-1Н-1,2,4-triazole [reaction (14)]. Special experiments revealed that thermal decomposition of compound 6 in 2 wt % in nitrobenzene at 93°С was significantly accelerated in the presence of nitrogen dioxide and upon addition of 5‒8% of 3-nitro-1Н-1,2,4-triazole, whereas the addition of sulfuric acid (5 wt % with respect to the solution, d = 1.84 g/cm3) significantly suppressed the autocatalytic nature of the process. Unfortunately, the kinetic curves in the latter case were poorly reproducible.
In conclusion, the reactions series (4)‒(17) (Scheme 3) with reaction (4) as the rate-limiting stage reasonably explained the effects of the azole concentration in the solution on the rate constant, activation parameters, negative value of the activation entropy of the limiting stage, kinetic features of the decomposition below the melting point, in the melt, and in the solution in dibutyl phthalate, autocatalysis, and the rate constant dependence on рKа.
Equation (1) could be used for the estimation of thermal stability of new 1-(dinitromethyl)-3-R-1Н-1,2,4-triazoles during their synthesis. Thermal stability of 1-(dinitromethyl)-3-nitro-1Н-1,2,4-triazole in the solution in dibutyl phthalate at 120°С was 23 times lower than that of 1-(1,1-dinitroethyl)-3-nitro-1Н-1,2,4-triazole [7], which pointed at higher risk associated with handling of 3-substituted 1-(dinitromethyl)-1H-1,2,4-triazoles during synthesis and storage.
EXPERIMENTAL
Compounds 1‒6 synthesized and purified as described elsewhere [1] contained at least 99% of the major component (chromatography data). Dibutyl phthalate used as solvent was washed three times with sodium hydrocarbonate solution, then washed with water to рН = 7.0, dried over magnesium sulfate, and distilled under vacuum. Nitrobenzene was purified by vacuum distillation and was found pure by chromatography, nD20 1.5527. The solvents were stored over silica gel in a vacuum dessicator.
Kinetics of thermal decomposition was studied by manometry method under isothermal conditions at residual air pressure 10–2‒10–1 mmHg using a Bourdon manometer [12]. Temperature of the thermostat was set with accuracy of ±0.2°. Differential thermal analysis was performed using a Q-1500 Paulik‒Paulik‒Erdey derivatograph over 20‒120°С at heating rate 2.5 deg/min [12]. The specimen mass was 50 mg. Intermediate and final gaseous decomposition products were identified by means of GLC using an LKhM-80 instrument (column with polysorb-1, length 12 m, temperature of evaporator and detector 40°С, detector current 150 mA) and IR spectroscopy using an UR-20 instrument (NaCl gas cell). Analysis of the condensed decomposition products was performed using a MKh-1312 chromato–mass spectrometer (temperature of the column with squalane 80°С, temperature of effusion chamber 100°С). The rate constants were calculated from the initial rate of thermal decomposition which was determined by differentiation of the kinetic curve. The accuracy of the rate constant determination was of ±6.5%. Standard deviation of the activation energy did not exceed 2.1 kJ/mol, and that of the logarithm of pre-exponential factor was of 0.22 un.
REFERENCES
Kofman, T.P., Trubitsyn, A.E., Dmitrienko, I.V., Glazkova, E.Yu., and Tselinskii, I.V., Russ. J. Org. Chem., 2007, vol. 43, no. 5, p. 758. https://doi.org/10.1134/S1070428007050193
Manelis, G.B., Nazin, G.M., and Prokudin, V.G., Doklady Phys. Chem., 2006, vol. 411, no. 5, p. 335. https://doi.org/10.1134/S0012501606120049
Manelis, G.B., Problemy kinetiki elementarnykh khimicheskikh reaktsii (Problems of the Kinetics of Elementary Chemical Reactions), Moscow: Nauka, 1973.
Nazin, G.M. and Prokudin, V.G., Abstracts of Paper, Mater. III Vseros. konf. “Energeticheskie kondensirovannye sistemy” (Proc. Of All-Russian Conf.“Energy Condensed Systems”), Chernogolovka, 2006, p. 187.
Bon, S., Chemistry of the Solid State, Garner, W.E., Ed., London: Butterworth, 1955.
Zbarskii, V.L., Maksimov, Yu.Ya., and Orlova, E.Yu., Trudy MKhTI im. D.I. Mendeleeva, 1967, no. 53, p. 84.
Stepanov, R.S., Kruglyakova, L.A., and Аstakhov, А.М., Russ. J. Org. Chem., 2007, vol. 43, no. 3, p. 474. https://doi.org/10.1134/S1070428007030293
Stepanov, R.S., Kruglyakova, L.A., and Аstaсhov, А.М., Book of Abstracts, 38th Int. ICT-Conf., 2007, Karlsruhe, p. 62/1.
Nikitina, E.V., Starova, G.L., Frank-Kameneckaya, O.V., and Pevzner, M.S., Kristallografiya, 1982, vol. 27, no. 3, p. 485.
Bagal, L.I. and Pevzner, M.S., Chem. Heterocycl. Comрd., 1970, vol. 6, no. 4, p. 517. https://doi.org/10.1007/BF00478408
Denesh, I., Titration in Non Aqueous Media, Moscow: Mir, 1971.
Stepanov, R.S., Fiziko-khimicheskie ispytaniya vzryvchatykh veshchestv (Physico-Chemical Tests of Explosives), Krasnoyarsk: KPI, 1989.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Rights and permissions
About this article

Cite this article
Kruglyakova, L.A., Stepanov, R.S., Pekhotin, K.V. et al. Kinetics and Mechanism of Thermal Decomposition of 1-(Dinitromethyl)-3-R-1H-1,2,4-triazoles. Russ J Gen Chem 90, 782–786 (2020). https://doi.org/10.1134/S1070363220050035
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363220050035