
Abstract
The extraction properties of 2,3-bis(diphenylphosphinyl)buta-1,3-diene (L1) and 3,4-bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene (L2) with respect to f elements are studied for the extraction of microquantities of U(VI) and Th(IV) from HNO3 solutions with solutions of the extracting agents in 1,2-dichloroethane. The introduction of methyl groups into the 1,3-butadiene carbon skeleton of bis(phosphine oxide) increases the efficiency of U(VI) extraction. The coordination properties of bis(phosphine oxides) L1 and L2 are studied for the complexes with uranyl nitrate. The crystal structures of bis(phosphine oxide) L1·THF (CIF file CCDC no. 2107403) and its complexes with uranyl nitrate I and II (CIF files CCDC nos. 2107404 and 2107405, respectively) are confirmed by X-ray diffraction data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In spite of a rather long history of studying phosphorus oxides, increased interest in the synthesis of efficient and selective organophosphorus ligands to various metal ions has been observed in recent decades. The brief list of areas actively using mono-, di-, and polydentate phosphine oxides is as follows: bioinorganic chemistry [1], catalysis [2], production of new materials [3], separation and extraction of rare and scattered elements [4], and hydrometallurgy and decontamination of nuclear waste of electric power stations from radioactive elements [5]. Metal complexes with bis(phosphine oxides) demonstrating a richer structural topology than monophosphine oxides are of most interest. For example, metal-organic frameworks [6–9], macrocyclic compounds [10], and cage complexes [11] based on bis(phosphine oxides) were described.
The structures of the synthesized complexes mainly depend on the structure of the bis(phosphine oxide) used. As a rule, methylene bis(phosphine oxide) chelates metal ions to form six-membered complexes [12, 13]. In the case of the elongation of the bridge linking two phosphine oxide groups Ph2P(O)(CH2)nP(O)Ph2 (n = 2, 4, or 6), dimeric or polymeric complexes are usually formed [14–16].
Continuing the study of the coordination properties of alkylenebis(phosphine oxide) [17] and its analogs [18], we performed studies related to an attempt to reveal the influence of structural changes in the carbon skeletons of bis(phosphine oxides) on their coordination and extraction properties. The bis(phosphine oxide) ligands on the 1,3-alkadiene platform, 2,3-bis(diphenylphosphinyl)buta-1,3-diene (L1) [19] and 3,4-bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene (L2) [20], were chosen as objects of the study.
EXPERIMENTAL
Solvents СНСl3, СН2Сl2, and acetonitrile (reagent grade) were distilled over P2O5 for dehydration and stored over CaH2, and THF was distilled and stored over CaH2 and distilled off prior to use. Commercial 1,2-dichloroethane and ethanol were used as received.
1Н, 13С, and 31Р NMR spectra were recorded in CDCl3 on a Bruker Avance 400 spectrometer (400.13, 100.61, and 161.98 MHz, respectively). The Н1 and 31Р NMR spectra of the complex were recorded in DMF-d7. IR spectra were recorded on a VERTEX 70v Fourier-transform IR spectrometer (Germany). High-resolution mass spectra were detected on a Bruker micrOTOF II instrument using the electrospray ionization (ESI) method. Elemental analyses (C, H, N) were carried out on a CarloErba 1106 automated analyzer.
Synthesis of 3,4-bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene (L2) was carried out in an inert atmosphere. 2,5-Dimethyl-3-hexyne-2,5-diol (0.9 g, 6.4 mmol) was added to a suspension of NaH (0.31 g, 12.8 mmol) in THF (6 mL) with vigorous stirring, and after 5 min diphenylchlorophosphine (3.12 g, 14.1 mmol) was added dropwise maintaining the temperature of the reaction mixture at 20°С. After the cessation of gas release, the mixture was stirred at 20°С for 4 h. A precipitate was filtered off and washed with THF (2 × 3 mL), and Cu(OTf)2 (0.2 g, 0.7 mmol, 10 mol %) was added to the filtrate. After the formation of a yellow-orange solution, the mixture was left to stay for 48 h. Then CH2Cl2 (20 mL) was added, the reaction mixture was washed with a 25% solution of NH3 (2 × 3 mL) and water (2 × 5 mL), and dried over Na2SO4. The drying agent was filtered off and washed with CH2Cl2 (2 × 5 mL), and the filtrate was evaporated in vacuo. Chromatography on silica gel in a CHCl3–benzene–methanol (30 : 30 : 1) system gave 3,4-bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene (L2) (2.39 g, 72%), (5-chloro-2,5-dimethylhex-3-in-2-yl)diphenylphosphine oxide (L3) (0.21 g, 9%), and (2,5-dimethylhex-5-en-3-in-2-yl)diphenylphosphine oxide (L4) (0.14 g, 7%).
3,4-Bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene (L2). Colorless crystals, m.p. = 186–188°C [20]. 1H NMR (δ, ppm): 8.00–7.96 m (4H, o-H, Ph), 7.48–7.44 m (6H, o,p-H, Ph), 7.38–7.35 m (2H, p-H, Ph), 7.20–7.16 m (4H, m-H, Ph), 7.14–7.08 m (4H, m-H, Ph), 1.76 s (6H, 2CH3), 1.69 s (6H, 2CH3). 13C NMR (δС, ppm): 153.88 dd ((CH3)2C=C), 2JP,C = 8.02 Hz), 134.75 d (ipso-C, 1JP,C = 102.13 Hz), 134.20 d (ipso-C, 1JP,C = 99.21 Hz), 132.15 dd (p-C, Ph, 4JP,C = 11.68 Hz, 4JP,C = 10.21 Hz), 131.20 br.d. (o-C, Ph, 2JP,C = 31.36 Hz), 129.02 d ((CH3)2C=C, 1JP,C = 8.02 Hz), 128.38 d (m-C, Ph, 3JP,C = 10.95 Hz), 128.38 d (m-C, Ph, 3JP,C = 11.68 Hz), 25.30–25.23 m ((CH3)2C=C), 25.12–25.09 m ((CH3)2C=C). 31P NMR (δP, ppm): 27.60 s. IR (ν, cm–1): 3051 ν(CH, Ph); 2910, 1436 ν(CH3); 1608, 1589 ν(Ph); 1628 ν(C=C); 1162 ν(P=O). MS, m/z: 511.1950 [M + H]+ (calculated for C32H33O2P2: 511.1961).
(5-Chloro-2,5-dimethylhex-3-in-2-yl)diphenylphosphine oxide (L3). Colorless oil. 1H NMR (δ, ppm): 7.72–7.68 m (4H, o-H, Ph), 7.52–7.50 m (2H, p-H, Ph), 7.48–7.43 m (4H, m-H, Ph), 1.46 d (6H, 2CH3, 3JP,H = 6.0 Hz), 1.44 s (6H, 2CH3). 13C NMR (δС, ppm): 133.29 d (ipso-C, Ph, 1JP,C = 104.32 Hz), 131.74 d (p-C, Ph, 4JP,C = 2.91 Hz), 131.36 d (o-C, Ph, 2JP,C = 9.48 Hz), 128.25 d (m-C, Ph, 3JP,C = 11.67 Hz), 102.95 br.s. (C≡C), 101.93 br.s. (C≡C), 99.74 d (C–P, 1JP,C = 13.86 Hz), 73.57 d (C–Cl, 4JP,C = 5.11 Hz), 30.74 d (2CH3, 5JP,C = 2.91 Hz), 19.00 d (2CH3, 2JP,C = 5.84 Hz). 31P NMR (δP, ppm): 34.37 s. MS, m/z: 345.1170 [M + H]+ (calculated for C20H23ClOP: 345.1181).
(2,5-Dimethylhex-5-en-3-in-2-yl)diphenylphosphine oxide (L4). Colorless crystals, m.p. = 152–154°C. 1H NMR (δ, ppm): 7.70–7.65 m (4H, o-H, Ph), 7.47–7.39 m (8H, Ph), 5.46 s (1H, =C(H)H), 5.02 s (1H, =C(H)H), 1.84 s (3H, CH3(=CH2)), 1.36 d (6H, 2CH3, 3JP,H = 6.1 Hz). 13C NMR (δС, ppm): 136.17 d (CH2=C–CH3, 5JP,C = 7.29 Hz), 133.21 d (ipso-C, Ph, 1JP,C = 105.04 Hz), 131.18 s (o-C, Ph, 2JP,C = 9.48 Hz), 131.41 s,Footnote 1 Ph, 128.14 d (Ph, 3JP,C = 12.41 Hz), 117.12 d (CH2=C–CH3, 4JP,C = 3.65 Hz), 101.09 br.s (C≡C), 100.08 br.s (C≡C), 99.45 d (C–P, 1JP,C = 13.14 Hz), 23.00 d (CH2=C–CH3, 5JP,C = 5.84 Hz), 19.10 d (2CH3, 2JP,C = 5.83 Hz). 31P NMR (δP, ppm): 31.57 s. MS, m/z: 309.1403 [M + H]+ (calculated for C20H22OP: 309.1414).
Synthesis of 2,3-bis(diphenylpphosphinyl)-but-1,3-diene uranyl dinitrate (I). A solution of UO2(NO3)2· 6H2O (104 mg, 0.2 mmol) in acetonitrile (5 mL) was slowly added to a solution of phosphine oxide L1 (90.8 mg, 0.2 mmol) in chloroform (5 mL), and the reaction mixture was stirred for 1 h. The precipitated yellow crystals were filtered off, washed with acetonitrile (2 × 5 mL), recrystallized from a DMF–diethyl ether (1 : 1) mixture, and dried in a vacuum of 0.1 mmHg to a constant weight. The yield of complex I was 160 mg (80%), Тdecomp = 255–257°С. IR (KBr; ν, cm–1): 539, 582, 692, 730, 929 (UO2), 1122, 1145, 1161, 1273 (UO2), 1292, 1385, 1487 (NO3), 1516. 1Н NMR (400 MHz; DMF-d7; δ, ppm): 8.4–7.6 m (20Н, 4С6Н5), 6.87 d (2Н, 2JHH = 42 Hz, СН2=), 6.18 br.s. (2Н, СН2=). 13С NMR (100 MHz; DMF-d7; δ, ppm): 138.94 dd (1JPC = 95.0 Hz, 2JPC = 8.0 Hz, Р–С=), 136.38 br.s (Н2С=), 134.04 br.s (С6Н5), 132.57 d (3JPC = 9.7 Hz, С6Н5), 129.68 d (2JPC = 12.0 Hz, С6Н5). 31Р NMR (162 MHz; DMF-d7; δ, ppm): 46.44 br.s.
For С28Н24N2O10P2U | |||
Anal. calcd., % | С, 39.64 | Н, 2.85 | N, 3.30 |
Found, % | С, 39.40 | Н, 2.89 | N, 3.44 |
Synthesis of 2,3-bis(diphenylphosphinyl)-1,1,4,4-tetramethylbut-1,3-diene uranyl dinitrate (II). A solution of UO2(NO3)2⋅6H2O (104 mg, 0.2 mmol) in ethanol (5 mL) was carefully added onto the surface of a solution of phosphine oxide L2 (102 mg, 0.2 mmol) in DMF (5 mL). The light green crystals precipitated on the next day were filtered off, washed with diethyl ether, and dried in a vacuum of 0.1 mmHg to a constant weight. The yield of complex II was 150 mg (73%), Тdecomp = 320–322°С. IR (KBr; ν, cm–1): 545, 575, 699, 748, 932 (UO2), 1097, 1121, 1144, 1278 (UO2), 1296, 1385, 1439, 1484 (NO3), 1512, 1529.
For С32Н32N2O10P2U | |||
Anal. calcd., % | С, 42.21 | Н, 3.54 | N, 3.08 |
Found, % | С, 42.00 | Н, 3.54 | N, 3.10 |
Study of the extraction properties for compounds L1, L2, and L5. 1,2-Dichloroethane (reagent grade) was used as the organic solvent. Solutions of the extracting agents were prepared using exact weighed samples. The starting aqueous solutions of U(VI) and Th(IV) were prepared by the dissolution of the corresponding nitrates in water followed by the addition of HNO3 to a required concentration. The starting concentration of metal ions was 2 × 10–6 mol/L. Equal volumes of the organic and aqueous phases were stirred at room temperature on a rotary instrument with a rate of 60 rpm for 1 h, which is sufficient for establishing constant distribution coefficients.
The concentration of U(VI) and Th(IV) in the starting and equilibrium aqueous solutions was determined by mass spectrometry using sample ionization in inductively coupled plasma (ICP-MS) on an XSeriesII mass spectrometer (Thermo Scientific, United States) at the following operation parameters: generator output 1300 W, package of standard nickel cones, PolyCon concentric sprayer, quartz conic spraying chamber cooled to 3°С, plasma forming argon flow rate 13 L/min, auxiliary argon flow rate 0.9 L/min, argon flow rate in the sprayer 0.95 L/min, and analyzed sample flow rate 0.8 mL/min. The spectrometer was calibrated using single- and multicomponent standards (High-Purity Standards, United States). The concentration of U(VI) and Th(IV) in the equilibrium organic phase was determined as a difference between the concentrations in the starting and equilibrium aqueous solutions. The distribution coefficients of elements (D) were calculated as the ratio of their concentrations in the equilibrium organic and aqueous phases. An inaccuracy of distribution coefficient determination did not exceed 5%. The concentration of HNO3 in the equilibrium aqueous phase was determined by potentiometric titration with a standard solution of NaOH.
X-ray diffraction (XRD). Single crystals of compound L1·THF were obtained by recrystallization from THF, and complexes I and II were taken from the reaction mixture. Reflection intensities were measured on Bruker Apex II (L1·THF and I at 120 K) and Bruker Quest (II, 100 K) diffractometers using MoKα radiation (λ = 0.71073 Å). The structures were solved using the SHELXT algorithm [21] and refined in the full-matrix approximation against F2 (hkl). Non-hydrogen atoms were refined anisotropically. Positions of hydrogen atoms were found from geometric concepts and refined in the isotropic approximation by the riding model with Uiso(H) = 1.2Ueq(X). Single crystals of L1·THF are twins that were separated using the PLATON program [22] and refined using the BASF/HKLF 5 instructions. All calculations were performed using the SHELXL2014 [23] and OLEX2 [24] programs. The parameters of the crystals and refinement results are given in Table 1.
An additional crystallographic information for L1·THF, I, and II was deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 2107403 (L1·THF), 2107404 (I), and 2107405 (II); http://www.ccdc.cam.ac.uk/structures).
The surfaces of the Voronoi polyhedra were constructed and examined in the ToposPro structural topological program package [25].
RESULTS AND DISCUSSION
Compound I was synthesized using a previously described procedure [19]. The synthesis of 1,3-alkadiene II was based on the modified procedure [20]. 2,5-Dimethyl-3-hexyne-2,5-diol was introduced in the reaction with diphenylchlorophosphine in the presence of NaH and 10 mol % Cu(OTf)2 in THF at 20°С (Scheme 1).

Scheme 1 .
The target product (alkadiene L2) was synthesized and isolated by column chromatography in a yield of 72%. (5-Chloro-2,5-dimethylhex-3-in-2-yl)diphenylphosphine oxide (L3) and (2,5-dimethylhex-5-en-3-in-2-yl)diphenylphosphine oxide (L4) are formed as by-products in 9 and 7% yields, respectively. The structures of compounds L2–L4 were determined by 1Н, 13С, and 31P NMR spectroscopy and IR spectroscopy, and the compositions were determined using elemental analyses and mass spectrometry.
The extraction properties of compounds L1 and L2 with respect to f elements were studied for the extraction of microquantities of U(VI) and Th(IV) from HNO3 solutions with solutions of the extracting agents in 1,2-dichloroethane. 1,2-Bis(diphenylphosphinyl)ethane Ph2P(O)CH2CH2P(O)Ph2 (L5) served as the reference compound.
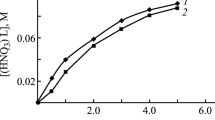
The influence of the HNO3 concentration in the equilibrium aqueous phase on the changes in the distribution coefficients of U(VI) (Fig. 1) and Th(IV) (Fig. 2) was considered for the extraction with 0.001 M solutions of compounds L1, L2, and L5 in dichloroethane. An increase in the concentration to 1–2 M HNO3 is accompanied by an increase in DU and DTh, which is related to the salting-out effect of \({\text{NO}}_{3}^{ - }\) ions and corresponds to the extraction of these ions as coordinatively solvated nitrates [26]. The further increase in the HNO3 concentration results in some decrease in DU and DTh due to the coextraction of nitric acid leading to the concentration of the free extracting agent in the organic phase.

Dependences of the distribution coefficients of U(VI) on the equilibrium concentration of HNO3 in the equilibrium aqueous phase for extraction with 0.001 M solutions of compounds L1, L2, and L5 in dichloroethane.

Dependences of the distribution coefficients of Th(IV) on the equilibrium concentration of HNO3 in the equilibrium aqueous phase for extraction with 0.001 M solutions of compounds L1, L2, and L5 in dichloroethane.
The stoichiometric metal to extracting agent ratio in the extracted complexes was determined by the equilibria shifting method. The obtained data (Figs. 3, 4) show that compounds L1, L2, and L5 in dichloroethane extract Th(IV) from nitric acid solutions in the form of disolvates, whereas U(VI) is predominantly extracted in the form of monosolvates.

Dependences of the distribution coefficients of U(VI) on the concentration of compounds L1, L2, and L5 in dichloroethane for extraction from a 3 M solution of HNO3.

Dependences of the distribution coefficients of Th(IV) on the concentration of compounds L1, L2, and L5 in dichloroethane for extraction from a 3 M solution of HNO3.
The data on the extraction of U(VI) and Th(IV) from nitric acid solutions under comparable conditions are given in Table 2 to compare the extraction abilities of compounds L1, L2, and L5 with respect to U(VI) and Th(IV) and the separation factors of thorium and uranium (βTh/U = DTh/DU). It can be seen that the introduction of allyl and especially methyl substituents into the alkylene bridge of the Ph2P(O)CH2CH2P(O)Ph2 (L5) molecule leads to an increase in the efficiency of U(VI) extraction and some decrease in the extraction ability of compounds L1 and L2 with respect to Th(IV).
The selectivity of Th(IV) extraction decreases in the series of compounds L5 > L1 > L2, and compound L2 extracts U(VI) much more efficiently than Th(IV). The separation factor of uranium and thorium (βU/Th) for this compound decreases from 200 to 16 with an increase in the HNO3 concentration in the equilibrium aqueous phase from 0.3 to 3 mol/L. The difference in the stoichiometry of the extracted U(VI) and Th(IV) complexes results in a decrease in βU/Th with an increase in the concentration of compound L2 in the organic phase.
In order to establish factors affecting the extraction properties of bis(phosphine oxides) L1 and L2, their structural studies were carried out. The structures and compositions of ligands L1 and L2 were determined by XRD. Bis(phosphine oxide) L2 crystallizes as monohydrate similarly to the earlier presented data [20], whereas compound L1 crystallizes with a THF molecule. The XRD data show that molecule L1 in the crystal of L1·THF adopts the planar s-trans conformation (with the С=С–С=С angle equal to 162.6(3)°) (Fig. 5), whereas L2 in L2·H2O [20] takes the gauche s‑cis conformation with the turning angle between the unsaturated fragments –С=С(СН3)2 equal to 86.5(2)°.

Molecular structure of ligand L1 in representation of atoms by thermal ellipsoids (p = 50%).
A violation of complanarity of the alkadiene fragment for compound L2 is probably induced by steric interactions between the terminal methyl groups with the bis(phosphine oxide) fragments. The coplanarity of the 1,3-alkadiene fragment in L1 favors the charge density delocalization along this chain, which results in some elongation of the C=C bonds (Table 2). In addition, the bonds of the phosphoryl group P=O and the bonds of phosphorus with the 1,3-alkadiene skeleton P–C are elongated in L1 compared to L2.
UV spectroscopy was used to establish the conformational compositions of compounds L1 and L2 in the liquid phase. It is known that electron spectroscopy is widely used to determine the conformational composition of flexible molecules of the 1,3-alkadiene type [27]. In particular, it was found that the violation of conjugation between the vinyl fragments of the carbon skeleton and the formation of the gauche s-cis conformation are accompanied by an appreciable decrease in the absorption band intensity in the UV spectra. This effect was demonstrated for organophosphorus 1,3-alkadienes [28]. Using this approach, we succeeded to show that the UV spectra of compounds L1 and L2 detected in solutions differ strongly. The intensity of the band at 220 nm for compound L1 is nearly 1.5 times higher than that for compound L2 (Fig. 6). Thus, it can be asserted with a high probability that the conformational compositions of bis(phosphine oxides) L1 and L2 in the solid phase and solution are identical.

UV spectra of (1) L1 and (2) L2.
The coordination properties of bis(phosphine oxides) L1 and L2 were studied for their reactions with UO2(NO3)2·6H2O (Scheme 2). Uranyl complexes I and II were synthesized using СН3СN as the solvent for ligand L1 and DMF for ligand L2.

Scheme 2 .
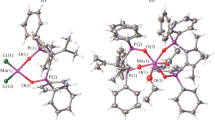
Ligands L1 and L2 form uranyl complexes in the ratio UO2 : L = 1 : 1, which was found by XRD of single crystals of complexes I and II. The molecular structures of the complexes are shown in Fig. 7. The coordination polyhedra U(VI)O8 are hexagonal bipyramids with the oxygen atoms of uranyl in the axial positions and six oxygen atoms in the equatorial positions. The uranyl groups in complexes I and II are nearly linear (177.2(1)° and 178.2(1)°, Table 3).

Molecular structures of compounds I and II in representation of atoms by thermal ellipsoids (p = 50%).
The U(VI)−OP=O bonds are shorter than the U(VI)−O(N) bonds. The bond lengths in bis(phosphine oxides) L3 and L4 are close to the bonds in the starting ligand L2·H2O [20]. The similarity of the conformations of the ligands for L2, I, and II compared to L1 is shown in Fig. 8.

Comparison of conformations of the ligands in the crystals of compounds (orange) L1·THF, (green) L2·H2O [20], (blue) I, and (red) II. The atoms of the P=O groups are superimposed. Hydrogen atoms are omitted.
As follows from Fig. 8, the configurations of the ligand in complexes I and II are very close to each other due to the rigidity of the seven-membered metallocycle, which assumes rotation only along the P–CPh bonds. The conformations of free phosphine oxide in L2·H2O [20] and in complex II are also close to each other on the whole, except for the turn of one Ph2P=O fragment along the P–Cdiene bond at the angle about 120°. At the same time, the configuration of molecule L1 in L1·THF differs considerably from the configuration in L3 because of the unusual torsion angles C=C–P=O. Thus, the s-trans conformation of ligand L1 can be a reason for its decreased extraction ability to f elements compared to its alkyl-substituted analogs, in particular, ligand L2.
Another possibility of different behaviors of ligands L1 and L2 during extraction can be related to different compositions of the atoms that form the molecular surface and, as a consequence, affecting the solubility of the substance in hydrophobic solvents. The surfaces of the molecular Voronoi polyhedra formed by all regions of the crystal, which are closer to this molecule than to the molecules (or ions) of the environment, provide a convenient approximation that makes it possible to estimate the contributions from diverse atoms to the surface area of the molecular surface and their participation in various intermolecular contacts [31]. This representation of molecules in crystal makes it possible to successfully analyze specific features of packing and intermolecular interactions in polymorphs [31–33], conformationally flexible molecules [33, 34], and homological series of compounds [35]. In particular, it has previously been found that an increase in the contribution of hydrophobic H…H contacts and C…H contacts to the total molecular surface area of monocarboxylic acids is accompanied by an increase in their ability to form polynuclear complexes with double uranium(VI) salts [35]. In the cases of L1·THF and L2·H2O, the surface area of the Voronoi molecule, which corresponds to hydrophobic interactions C…C, C–H…C, and C–H…H–C, is equal to 460 and 506 Å2 (or 87.5 and 91.7% of the total surface area of the molecule). In complexes I and II, the partial contribution of the hydrophobic interactions to the surface area of the molecule is 52.9 and 59.6% (327 and 387 Å2). Thus, as should be expected, alkyl-substituted dienes and their complexes should have a large molecular surface area capable of forming hydrophobic interactions.
The extraction and coordination properties of 2,3‑bis(diphenylphosphinyl)buta-1,3-diene (L1) and 3,4-bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene (L2) were studied in this work. Compound L1 was found by XRD and electron spectroscopy to have a predominantly planar s-cis conformation. The introduction of additional substituents as methyl groups (compound L2) into the 1,3-butafiene fragment leads to a gauche s-cis conformation with the turning angle between the unsaturated fragments С=С(СН3)2 equal to 86.5(2)°. A similar conformation of the ligands was determined for uranyl complexes I and II. The study of the extraction properties of bis(phosphine oxides) L1 and L2 revealed a significant efficiency of extracting agent L2 compared to compound L1 for the extraction of microquantities of U(VI) from HNO3 solutions. Probably, this can be explained by an enhanced hydrophobicity of bis(phosphine oxide) L2 and uranyl complex II and a more favorable conformation of ligand L2 for the formation of a complex with U(VI).
Notes
The signal of the left part of the p-C coalesces with the left part of the o-C doublet.
REFERENCES
Demkowicz, S., Rachon, J., Daśko, M., and Kozak, W., RSC Adv., 2016, vol. 6, p. 7101.
Ni, H., Chan, W.-L., and Lu, Y., Chem. Rev., 2018, vol. 118, p. 9344.
Baumgartner, T. and Réau, R., Chem. Rev., 2006, vol. 106, p. 4681.
Rozen, A.M. and Krupnov, B.V., Russ. Chem. Rev., 1996, vol. 65, p. 973.
Myasoedov, B.F. and Kalmykov, S.N., Mendeleev Commun., 2015, vol. 25, no. 5, p. 319.
Mathew, M. and Palenik, G.J., Inorg. Chim. Acta, 1971, vol. 5, p. 573.
Harrison, P.G., Sharpe, N.W., Pelizzi, C., et al., Dalton Trans., 1983, p. 921.
Pettinari, C., Marchetti, F., Cingolani, A., et al., Inorg. Chim. Acta, 1991, vol. 312, p. 125.
Spichal, Z., Necas, M., and Pinkas, J., Inorg. Chem., 2005, vol. 44, p. 2074.
Al-Resayes, S.I., Hitchcock, P.B., and Nixon, J.F., Chem. Commun., 1991, p. 78.
Beagley, B., Dyer, G., McAuliffe, C.A., et al., Chem. Commun., 1991, p. 965.
Jin, Q.-H., Wu, J.-Q., Zhang, Y.-Y., et al., NCS, 2009, vol. 224, p. 428.
Lees, A.M.J. and Platt, A.W.G., Inorg. Chem., 2003, vol. 42, p. 4673.
Spichal, Z., Necas, M., and Pinkas, J., Inorg. Chem., 2005, vol. 44, p. 2070.
Spichal, Z., Petricek, V., Pinkas, J., and Necas, M., Polyhedron, 2008, vol. 27, p. 283.
Spichal, Z., Necas, M., Pinkas, J., and Zdrahal, Z., Polyhedron, 2006, vol. 25, p. 2006.
Berezin, A.S., Davydova, M.P., Bagryanskaya, I.Yu., et al., Inorg. Chem. Commun., 2019, vol. 107, p. 107473.
Artem’ev, A.V., Davydova, M.P., et al., Dalton Trans., 2019, vol. 48, no. 43, p. 16448.
Pollok, T. and Schmidbaur, H., Tetrahedron Lett., 1987, vol. 28, p. 1085.
Chen, F., Xia, Y., Lin, R., et al., Org. Lett., 2019, vol. 21, p. 579.
Sheldrick, G.M., Acta Crystrallogr., Sect. A: Found. Adv., 2015, vol. 71, p. 3.
Spek, A.L., Acta Crystrallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 9.
Sheldrick, G.M., Acta Crystrallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Blatov, V.A., Shevchenko, A.P., and Proserpio, D.M., Cryst. Growth Des., 2014, vol. 14, p. 3576.
Turanov, A.N., Karandashev, V.K., Kharitonov, A.V., et al., Solvent Extr. Ion Exch., 2000, vol. 18, p. 1109.
Gillam, A.E., Stern, E.S., and Timmons, C.J., Gillam and Stern’s Introduction to Elektronic Absorption Spectroskopy in Organic Chemistry, London: Edward Arnold, 1970.
Ratovskii, G.V., Sergienko, L.M., Brel, V.K., et al., Zh. Obshch. Khim., 1983, vol. 53, p. 1186.
Berezin, A.S., Samsonenko, D.G., Brel, V., and Artem’ev, A.V., Dalton Trans., 2018, vol. 47, p. 7306.
Davydova, M.P., Bauer, I.A., Brel, V.K., et al., Eur. J. Inorg. Chem., 2020, p. 695.
Serezhkina, L.B. and Vologzhanina, A.V., Acta Crystallogr., Sect. B: Struct. Sci. Cryst. Eng. Mater., 2012, vol. 68, p. 305.
Zerezhkin, A.N. and Savchenkov, A.V., Cryst. Growth Des., 2015, vol. 15, p. 2878.
Zerezhkin, V.N. and Savchenkov, A.V., Cryst. Growth Des., 2020, vol. 20, p. 1997.
Vologzhanin, A.V., Ushakov, I.E., and Korlyukov, A.A., Int. J. Mol. Sci., 2021, vol. 21, p. 8970.
Savchenkov, A.V., Klepov, V.V., Vologzhanina, A.B., et al., CrystEngComm, 2015, vol. 17, p. 740.
ACKNOWLEDGMENTS
Elemental analyses and NMR, IR, and Raman spectra recording were supported by the Ministry of Science and Higher Education of the Russian Federation using scientific equipment of the Center for Molecular Structure Investigation at the Institute of Organoelement Compounds, Russian Academy of Sciences.
Funding
This work was supported by the Russian Science Foundation, project no. 20-13-00329.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Brel, V.K., Artyushin, O.I., Morgalyuk, V.P. et al. Extraction and Coordination Properties of 2,3-Bis(diphenylphosphinyl)buta-1,3-diene and 3,4-Bis(diphenylphosphinyl)-2,5-dimethylhexa-2,4-diene. Russ J Coord Chem 48, 201–209 (2022). https://doi.org/10.1134/S1070328422040017
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328422040017