
Abstract
The reaction of new 2,6-bis(pyrazol-3-yl)pyridine ligand (L) containing a redox-active ferrocenyl substituent with cobalt(II) salt gave the cobalt(II) complex [Co(L)2](ClO4)2 (I), which was isolated in a pure state and characterized by elemental analysis, NMR spectroscopy, cyclic voltammetry, and X-ray diffraction. According to X-ray diffraction data (CIF file CCDC no. 2049714) and the Evans method, which allows determining the spin state of paramagnetic compounds in solution on the basis of NMR spectra, the cobalt(II) ion in complex I occurs in the high-spin state and does not undergo temperature-induced spin transition in the temperature range of 120–370 K.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Some transition metal complexes can switch between two spin states under an appropriate external stimulus [1, 2], e.g., under the action of temperature or pressure, irradiation with light, or application of a magnetic field. A spin transition of this type is accompanied by significant changes in the magnetic and optical properties, which determine the prospects for practical application of these complexes in displays [3], electroluminescent devices [4], molecular switches [5], chemical sensors [6, 7], etc.
Among the wide diversity of compounds able to undergo a spin transition (most often, under the action of temperature), this ability is most often found in iron(II) and cobalt(II) complexes in the (pseudo)octahedral environment of nitrogen-containing ligands [2]. However, all applications indicated above require control over the spin state of the metal ion, which is attained by some chemical modification of the ligands [8]. 2,6-Bis(pyrazol-1-yl)pyridines [9] and isomeric 2,6-bis(pyrazol-3-yl)pyridines [10] are among the most popular classes of organic ligands to be used for this purpose due to extensive opportunities of their chemical functionalization, which allow for real [8] molecular design of 2,6-bis(pyrazol-1-yl)pyridine [11] and 2,6-bis(pyrazol-3-yl)pyridine [12] complexes with specified parameters of temperature-induced spin transition by choosing substituents with desired electronic properties.
One more method for control of the spin state of transition metal complexes is decoration of the ligand with pH-sensitive groups that change electromeric characteristics upon protonation/deprotonation and thus affect the spin state of the metal ion [13]. The same goal can be attained by using redox-active substituents such as ferrocene [14], which is itself diamagnetic. However, redox reactions of ferrocene can reversibly switch the electronic structure of the ligand, which is used in catalysis [15]. The introduction of redox-active ferrocenyl substituent to the bis(pyrazolyl)pyridine ligand can considerably expand the possibilities of controlling the metal ion spin state by modification of bis(pyrazolyl)pyridine and can give the first complex capable of redox switching and temperature-induced spin transition.
Here we synthesized a new 2,6-bis(pyrazol-3-yl)pyridine (L) containing a ferrocenyl group in position 5 of the pyrazol-3-yl ring (Scheme 1). Mixing of this ligand with cobalt(II) perchlorate as a source of transition metal ion gave the complex [Co(L)2](ClO4)2 (I) in a high yield.

Scheme 1 .
The spin state of the cobalt(II) ion in complex I was established using single crystal X-ray diffraction study at 120 K. The possibility of temperature-induced spin transition in solution in the 230–370 K range was studied using the Evans method traditionally used for this purpose, which is based on readily available NMR spectroscopy.
EXPERIMENTAL
All operations related to the synthesis of the bis(pyrazol-3-yl)pyridine ligand and the complex were performed in air using commercially available organic solvents distilled under argon. Co(ClO4)2⋅ 6H2O (Sigma-Aldrich) was used as received. Esterification of pyridine-2,6-dicarboxylic acid (Acros) with ethanol was carried out by a reported procedure [16] in the presence of sulfuric acid. Analysis for carbon, nitrogen, and hydrogen was performed on a Carlo Erba model 1106 analyzer.
Synthesis of 2,6-bis(5-ferrocenyl-1H-pyrazol-3-yl)pyridine (L). A solution of diethyl pyridine-2,6-dicarboxylate (2.80 g, 12.5 mmol) in dry THF was mixed with NaH (1.22 g, 30.5 mmol, 50% mineral oil suspension). A solution of acetylferrocene (6.27 g, 27.5 mmol) in THF (50 mL) was added dropwise to the resulting suspension. The reaction mixture was refluxed for 4 h and concentrated, and the solid residue was washed with hot hexane (2 × 30 mL), dried in a vacuum and dispersed in water; pH of the resulting suspension was brought to 5 by adding 1 M HCl. The violet precipitate was collected on a filter, dried in vacuum, and dispersed in ethanol with the addition of hydrazine hydrate (1.5 mL, 40 mmol). The reaction mixture was refluxed for 4 h. The precipitate that formed was collected on a filter, washed with ethanol, and dried in a vacuum. The yield was 2.5 g (35%).
1H NMR (DMSO-d6; 400 MHz; δ, ppm): 4.09 (s, 5H, Fc), 4.35 (s, 2H, Fc), 4.80 (s, 2H, Fc), 7.14 (s, 2H, Pz), 7.88 (s, 3H, Py), 13.02 (s, 2H, NH).
For C31H25N5Fe2 | |||
Anal. calcd., % | C, 64.28 | H, 4.35 | N, 12.09 |
Found, % | C, 64.31 | H, 4.40 | N, 12.17 |
Synthesis of [Co(L)2](ClO4)2 (I). Iron perchlorate hexahydrate (0.0365 g, 0.1 mmol) and 2,6-bis(5-ferrocenyl-1H-pyrazol-3-yl)pyridine (0.116 g, 0.2 mmol) were stirred in acetonitrile for 3 h. For purification, the resulting solution was concentrated, and diethyl ether was added dropwise until a precipitate formed. The mixture was kept for 12 h at −10°C. The precipitate was collected on a filter and dried in a vacuum. The yield was 126 mg (91%). 1H NMR (DMF-d7; 300 MHz; δ, ppm): 8.12 (br.s, 20H, Fc), 8.16 (br.s, 8H, Fc), 10.49 (br.s, 8H, Fc), 33.41 (br.s, 2H, p-Py−H), 53.90 (br.s, 4H, pyraz−CH), 83.72 (br.s, 4H, NH).
For C62H50N10O8Cl2Fe4Co | |||
Anal. calcd., % | C, 52.58 | H, 3.56 | N, 9.89 |
Found, % | C, 52.61 | H, 3.65 | N, 9.84 |
The single crystals of complex I suitable for X-ray diffraction were prepared by diffusion of diethyl ether vapor into its solution in methanol.
X-ray diffraction study of I was carried out on a Bruker APEX2 CCD diffractometer (MoKα radiation, graphite monochromator, ω-scan mode). The structure was solved using the ShelXT program [17] and refined by full-matrix least-squares calculations using the Olex2 program [18] in the anisotropic approximation on \(F_{{hkl}}^{2}\). The hydrogen atom positions were calculated geometrically and refined in the isotropic approximation using the riding model. The disordered solvent molecules (methanol and diethyl ether) were described as a diffusive contribution to the total scattering using the Solvent Mask option of the Olex2 program [18]. Selected crystallographic data and refinement parameters are given in Table 1.
The structural data for complex I were deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2049714; http://www.ccdc.cam.ac.uk/).
1H NMR spectra were recorded in DMSO-d6 and DMF-d7 on Bruker Avance 300 and 400 spectrometers operating at 300.15 and 400 MHz for protons, respectively. The chemical shifts (δ, ppm) were referred to the residual solvent signal (1H at 2.5 ppm for DMSO-d6 and 1H at 8.03 ppm for DMF-d7) or to the signal of 1% Me4Si admixture (1H at 0.0 ppm). The following parameters were used: spectral range of 120 ppm, recording time of 0.1 s, relaxation delay of 0.1 s, pulse duration of 9.5 μs, and number of acquisitions of 64. For increasing the signal-to-noise ratio, the obtained free induction decays were processed using exponential weighting with a coefficient of up to 3.
The temperature dependence of the magnetic susceptibility of complex I was determined by the Evans method [19] in DMF-d7 in the 230–370 K temperature range using an NMR tube with a coaxial insert. The inner (control) tube was filled with DMF-d7 with addition of ~1% Me4Si, while the outer tube contained a solution of the paramagnetic complex (~1–5 mg/cm3) in DMF-d7 with the same Me4Si concentration. The molar magnetic susceptibility was calculated from the difference between the Me4Si chemical shift in pure DMF-d7 and in a DMF-d7 solution of the complex (Δδ, Hz) by the following equation:
M is the molar mass of the complex, g/mol; ν0 is the spectrometer frequency, Hz; Sf is the magnetic form factor (4π/3); c is the concentration of the complex, g/cm3; \({{\chi }}_{{\text{M}}}^{{{\text{dia}}}}\) is the molar diamagnetic contribution to paramagnetic susceptibility calculated using Pascal’s constants [20]. The concentration c was calculated for each temperature according to the change in the solvent density (ρ): cT = mcompρ/msoln, where mcompρ is the weight of the complex, and msoln is the solution weight.
Complex I was also studied by cyclic voltammetry (CV). Cyclic voltammograms were measured on an Autolab PGSTAT128N potentiostat galvanostat operated by NOVA 2.0 software in a standard three-electrode cell. A 3 mm glassy carbon working electrode, a platinum wire counter electrode, and an Ag/AgCl reference electrode in a saturated KCl solution were used. The required conductivity of the solution was attained by adding TBAPF6 to the test solution up to a 0.1 M concentration. All CV curves were measured at a potential sweep rate of 100 mV/s. Before each experiment, the test solution was purged with argon for 5 min to remove the dissolved air, and the working electrode was thoroughly polished. All measurements were carried out at room temperature.
RESULTS AND DISCUSSION
For the synthesis of L (Scheme 2), the starting diethyl 2,6-pyridinedicarboxylate was introduced into the Claisen condensation with acetylferrocene under the action of sodium hydride in THF. The product thus formed was used, without purification, for the subsequent condensation with hydrazine hydrate in ethyl alcohol, which afforded the target ligand L in a moderate yield. This compound was poorly soluble even in solvents such as DMSO or DMF; therefore, it precipitated from the solution during the reaction and did not require further purification.

Scheme 2 .
The subsequent reaction of ligand L with cobalt perchlorate hydrate as a source of cobalt(II) in acetonitrile gave the homoleptic complex [Co(L)2](ClO4)2 (I) in a high yield. The complex was isolated in a pure state and characterized by elemental analysis, NMR spectroscopy, and CV. According to CV measurements (Fig. 1), an acetonitrile solution of complex I containing tetrabutylammonium hexafluorophosphate as a supporting electrolyte exhibited two reversible single-electron oxidation waves with E1/2 = ‒1.066 and 0.654 V versus Ag/Ag+ pair. The first process corresponds to the Co2+/Co+ oxidation of the cobalt ion, while the second oxidation potential is close to the potential of the Fc/Fc+ pair. Since the distances between oxidation–reduction half-waves (ΔEp = Epa – Epc) were 80 mV, both processes were classified as Nernst processes. The ΔEp value for the reference Fc/Fc+ pair was also 80 mV under the same conditions [21].

Cyclic voltammogram of a 1 mM solution of complex I in acetonitrile, C(TBAPF6) = 0.1 M, potential sweep rate: 100 mV/s, glassy carbon electrode (3 mm).
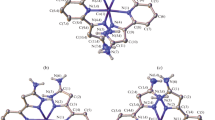
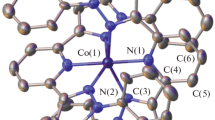
To confirm the results on the composition and structure of complex I obtained in this way, the complex was studied by single crystal X-ray diffraction (Fig. 2). The crystals were formed as crystal solvates with disordered solvent molecules upon diffusion of diethyl ether vapor into a solution of I in methanol (see Experimental). This allowed determination of the cobalt(II) spin state, which proved to be high-spin at 120 K, as indicated by the Co–N distances with the nitrogen atoms of bis(pyrazol-3-yl)pyridine ligands (C.N. 6) (Table 2), typical of high-spin cobalt(II) complexes (2.0–2.2 Å [2]). Iron(II) ions of the ferrocenyl substituents occur, as expected, in the low-spin (diamagnetic) state observed for ferrocene and its derivatives.

General view of complex I. Here and below, the perchlorate anions and hydrogen atoms (except for those belonging to NH groups) are omitted, and the rest of the atoms are presented as thermal vibration ellipsoids (p = 50%). Numbering is given only for metal ions and selected heteroatoms.
The bis(pyrazol-3-yl)pyridine ligands in complex I form a nearly octahedral coordination environment around the central metal ion (Fig. 3). For example, the θ angle between the root-mean-square planes of two ligands and the N(Py)MN(Py) angle, which are 90° and 180° for a perfect octahedron, amount to 88.1127(13)° and 178.1428(7)°. The shape of the coordination polyhedron CoN6 is characterized by the so-called “shape measures” [22], which describe its deviation from the perfect octahedron S(OC-6). The smaller this value, the better the coordination polyhedron shape is described by the corresponding perfect polyhedron. In complex I, the octahedral “shape measure” S(OC-6), estimated from X-ray diffraction data using the Shape 2.1 program [22], proved to be 4.021 (Table 2). For comparison, the “shape measure” characterizing the deviation from one more six-vertex perfect polyhedron, trigonal prism (TPR-6), which is often encountered in high-spin iron(II) complexes with N-heterocycles [23], is markedly higher (11.012) (Table 2).

Projection of complex I in the perpendicular direction illustrating a nearly octahedral shape of the cobalt(II) coordination polyhedron. Numbering is given only for metal ions.
The slight deviation of the shape of cobalt(II) coordination polyhedron in complex I from the perfect octahedron is caused, first of all, by the rigidity of the bis(pyrazol-3-yl)pyridine ligand [24]. For example, similar “shape measures” were observed previously for high-spin cobalt(II) complexes with other bis(pyrazol-3-yl)pyridines [25, 26]. However, a steric effect of the bulky ferrocenyl substituents in position 5 of the pyrazol-3-yl ring, which are located perpendicular to the ring plane, cannot be ruled out either (Table 2). The indicated substituents are rotated in pairs to face each other (Fig. 3). This arrangement gives rise to infinite chains of the [Co(L)2]2+ cations (Fig. 4) arranged along the crystallographic b axis and formed via stacking interactions between the ferrocene cyclopentadienyl rings and pyrazol-3-yl moieties connected to them, with the angle between them being 6.0(5)°–7.9(5)° and the distance between their centroids being 3.521(7)–3.828(7) Å. They are additionally stabilized by shortened C–H…N contacts (C…N 3.410(16) Å, CHN 159.3(6)°) between the same cyclopentadienyl rings and pyrazol-3-yl moieties of the neighboring [Co(L)2]2+ cations perpendicular to them and by the C–H…O contacts with the perchlorate anions (H…O 2.4361(9)–2.9097(7) Å), which “frame” the above-mentioned infinite chains. Thus, the cobalt(II) ion surrounded by the bis(pyrazol-3-yl)pyridine ligands with bulky electron-donating ferrocenyl substituents located in positions 5 of the pyrazol-3-yl rings of complex I occurs in a high-spin state in the crystal at 120 K, as unambiguously follows from X-ray diffraction data at this temperature.

Fragment of the crystal packing of complex I illustrating the formation of infinite chains in the crystal due to stacking interactions.
According to the results of the Evans method [19], based on the use of NMR spectroscopy, cobalt(II) remains in the same spin state in a solution of complex I in DMF. The choice of this solvent allowed us to record NMR spectra in a wide temperature range (230–370 K). The Evans method is actively used to search for new metal complexes with a temperature-induced spin transition [8] as the most easily accessible way to measure the magnetic susceptibility of solutions. According to this method, a special coaxial insert is placed into an NMR tube containing a solution of the paramagnetic complex and a standard compound such as tetramethylsilane (TMS) in a known concentration. The insert is filled with a TMS solution in the same solvent, in which no precipitation would take place on cooling; this is a limitation of the Evans method. The difference between the TMS chemical shifts in the NMR spectra recorded for these two solutions can be used to calculate the magnetic susceptibility of the solution of the paramagnetic compound and thus unambiguously determine the spin state of the metal ion. For the solution of complex I in DMF, χT value calculated in this way (Fig. 5) virtually does not deviate from the value of 3.9 cm3 mol–1 K, which corresponds to high-spin (S = 3/2) cobalt(II) over the whole temperature range 230–370 K accessible for the given solvent. The observed stabilization of the high-spin state in solution in which there are no crystal packing effects [8] attests to the “intramolecular” nature of this phenomenon.

Temperature dependence of the magnetic susceptibility of complex I in DMF according to the Evans method of NMR spectroscopy.
Thus, we synthesized and characterized the cobalt(II) complex with a new 2,6-bis(pyrazol-3-yl)pyridine ligand containing a redox-active ferrocenyl substituent in position 5 of the pyrazol-3-yl ring. According to the results of a low-temperature X-ray diffraction study of the complex, first of all, the Co–N bond lengths, the cobalt(II) ion in this complex occurs in the high-spin state (S = 3/2) even at 120 K. It does not undergo a temperature-induced spin transition in DMF, which is confirmed by NMR spectroscopy data (Evans method) in the temperature range of 230–370 K. Presumably, the replacement of the cobalt(II) ion by an iron(II) ion, which can change the spin state under the action of temperature when occurs in the 2,6-bis(pyrazol-3-yl)pyridine ligand environment [12], would result in the synthesis of the first 2,6-bis(pyrazol-3-yl)pyridine complex combining the redox-switching properties and temperature-induced spin transition.
REFERENCES
Sato, O., Tao, J., and Zhang, Y.-Z., Angew. Chem., Int. Ed. Engl., 2007, vol. 46, no. 13, p. 2152.
Spin-Crossover Materials: Properties and Applications, Halcrow, M.A., Ed., Chichester: Wiley, 2013.
Kahn, O. and Martinez, C.J., Science, 1998, vol. 279, no. 5347, p. 44.
Matsuda, M., Isozaki, H., and Tajima, H., Thin Solid Films, 2008, vol. 517, no. 4, p. 1465.
Senthil Kumar, K. and Ruben, M., Coord. Chem. Rev., 2017, vol. 346, p. 176.
Coronado, E. Gimenez-Marques, M., et al., J. Am. Chem. Soc., 2013, vol. 135, no. 43, p. 15986.
Bartual-Murgui, C., Akou, A., Thibault, C., et al., J. Mat. Chem., 2015, vol. 3, no. 6, p. 1277.
Halcrow, M.A., Crystals, 2016, vol. 6, no. 5, p. 58.
Halcrow, M.A., Coord. Chem. Rev., 2009, vol. 253, no. 21, p. 2493.
Halcrow, M.A., Coord. Chem. Rev., 2005, vol. 249, no. 25, p. 2880.
Kershaw-Cook, L.J., Kulmaczewski, R., Mohammed, R., et al., Angew. Chem., Int. Ed. Engl., 2016, vol. 55, no. 13, p. 4327.
Nikovskiy, I., Polezhaev, A.V., Novikov, V.V., et al., Chem.-Eur. J., 2020, vol. 26, p. 5629.
Gaudette, A.I., Thorarinsdottir, A.E., and Harris, T.D., Chem. Commun., 2017, vol. 53, no. 96, p. 12962.
Romero-Morcillo, T., Valverde-Munoz, F.J., Pineiro-Lopez, L., et al., Dalton Trans., 2015, vol. 44, no. 43, p. 18911.
Luca, O.R. and Crabtree, R.H., Chem. Soc. Rev., 2013, vol. 42, no. 4, p. 1440.
Polezhaev, A.V., Chen, C.-H., Kinne, A.S., et al., Inorg. Chem., 2017, vol. 56, no. 16, p. 9505.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, p. 112.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Evans, D.F., J. Chem. Soc., 1959, p. 2003.
Bain, G.A. and Berry, J.F., J. Chem. Educ., 2008, vol. 85, no. 4, p. 532.
Bligh, R.Q., Moulton, R., Bard, A.J., et al., Inorg. Chem., 1989, vol. 28, no. 13, p. 2652.
Alvarez, S., Chem. Rev., 2015, vol. 115, p. 13447.
Kershaw Cook, L., Mohammed, R., and Sherborne, G., Coord. Chem. Rev., 2015, vols. 289–290, p. 2.
Alvarez, S., J. Am. Chem. Soc., 2003, vol. 125, no. 22, p. 6795.
Pavlov, A.A., Belov, A.S., Savkina, S.A., et al., Russ. J. Coord. Chem., 2018, vol. 44, no. 8, p. 489. https://doi.org/10.1134/S1070328418080067
Pavlov, A.A., Nikovskii, I.A., Polezhaev, A.V., et al., Russ. J. Coord. Chem., 2019, vol. 45, no. 6, p. 402. https://doi.org/10.1134/S1070328419060046
ACKNOWLEDGMENTS
X-ray diffraction studies were performed using research equipment of the Center for Studies of Molecular Structure of the Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, and were supported by the Ministry of Science and Higher Education of the Russian Federation.
Funding
This study was supported by the Russian Science Foundation (grant no. 17-13-01456).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by Z. Svitanko
Rights and permissions
About this article

Cite this article
Denisov, G.L., Nikovskii, I.A., Aliev, T.M. et al. Spin State of Cobalt(II) 2,6-Bis(pyrazol-3-yl)pyridine Complex with a Redox-Active Ferrocenyl Substituent. Russ J Coord Chem 47, 480–487 (2021). https://doi.org/10.1134/S1070328421060014
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328421060014