
Abstract
Three new binuclear aqua-bridged nickel(II) 2-methylpropionate complexes [Ni2(μ-OH2)(μ-O2CCH(CH3)2)2L2–4((CH3)2CHCO2)2] (L is N,N,N',N'-tetramethylethylenediamine (I), pyridine (II), and 2,2'-bipyridyl (III)) are synthesized. The complexes are characterized by elemental and thermal analyses, mass spectrometry, IR and UV–Vis spectroscopy, and X-ray structure analysis (CIF files CCDC nos. 1840763 (I), 1913469 (II), and 1913471 (III)). The forms in which the synthesized coordination compounds exist in acetonitrile solutions are proposed on the basis of analyzing their mass spectra.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Transition metal complexes containing several coordination metal centers attract attention due to specific catalytic [1–3], magnetic [4–7], and spectral [8, 9] properties as well as a possible use in pharmacology [10, 11]. The binuclear complexes with bridging carboxylate ligands are among the most studied systems. A wide series of the binuclear nickel(II) complexes with aqua-, hydroxo-, or phenolate-bridging ligands [12–14] is interesting because these complexes resemble the active center the urease enzyme [15–18]. To the present time, several tens of the nickel(II) compounds with the [Ni2(µ-OH2)(µ-O2CR)2L2]2+ framework (L is the bidentate aminate ligand) were deposited with the Cambridge Crystallographic Data Centre (version 5.40, February 2019 [19]) [20–31]. When considering similar compounds as catalysts for homogeneous processes, it is important to study specific features of their behavior in solutions. However, this problem is insufficiently presented in the literature.
In this work, we studied the influence of the nature of the N-donor ligands in the binuclear nickel(II) complexes [Ni2(μ-OH2)(μ-O2CCH(CH3)2)2L2–4-((CH3)2CHCO2)2] (L is N,N,N',N'-tetramethylethylenediamine (Tmeda for I), pyridine (Рy for II), and 2,2'-bipyridyl (Вipy for III)) on the structures of the complexes, the character of thermal decomposition, and specific features of fragmentation in acetonitrile solutions.
EXPERIMENTAL
All compounds and solvents were purchased from commercial sources and used as received. Synthetic hellyerite NiCO3 ∙ 5.5H2O was obtained using the described procedure [32].
IR absorption spectra were recorded in a range of 400–4000 cm–1 on a Shimadzu IRTracer-100 instrument equipped with a Specac Quest ATR accessory for attenuated total internal reflection (ATR). Electronic absorption spectra were measured on an SF-56 instrument using quartz cells (l = 1 cm). Elemental analyses for C, H, and N were carried out using a LECO CHNS(O)-932 analyzer.
Mass spectrometric analyses (ESI-MS) of solutions of the complexes were carried out on a TSQ Quantum Access Max instrument (Thermo Fisher Scientific). Solutions of the samples (0.1 mg/mL) were introduced directly into electrospray with a flow rate of 10 μL/min. The voltages on the sprayer and capillary were ±5 kV and ±5 V, respectively. The pressure of the carrier gas (dry nitrogen) was 34.5 kPa, and the temperatures of the evaporator and capillary were 70 and 200°С, respectively. The spectra were recorded in the range of most intense signals: from 100 to 1000 Da.
The thermal decomposition of complexes I–III was studied by thermogravimetry (TG) on a Shimadzu DTG-60 analyzer. Experiments were carried out in dry nitrogen with a constant flow rate of 10 deg/min. The studied samples were heated in open aluminum crucibles, and the weight of each individual sample did not exceed 10 mg.
Synthesis of μ-aqua-k2O:O-di-μ-2-methylpropionato-k4O:O'-bis[(2-methylpropionato-kO)(N,N,N',N'- tetramethylethylenediamine-k2N,N')nickel(II)][Ni2(μ-OH2)(μ-O2CCH(CH3)2)2(Тmeda)2((CH3)2-CHCO2)2] (I). 2-Methylpropionic acid (0.56 mL, 6 mmol) was added to a suspension of NiCO3 ∙ 5.5H2O (0.6534 g, 3 mmol) in an acetonitrile–water (100 : 1) mixture (40 mL), and a solution of Tmeda (0.45 mL, 3 mmol) in acetonitrile (10 mL) was poured to the reaction mixture. Green crystals suitable for X-ray structure analysis precipitated from the mother liquor during slow evaporation in air. The yield of complex I was ~90%.
For C28H62N4O9Ni2 | |||
Anal. calcd., % | C, 47.04 | H, 8.75 | N, 7.84 |
Found, % | C, 47.21 | H, 8.50 | N, 7.93 |
IR (ν, cm–1): 3019 w, 2960 w, 2866 w, 2838 w, 2801 w, 2360 m, 2000 w, 1618 s, 1616 s, 1522 w, 1461 s, 1414 s, 1369 m, 1284 m, 1194 w, 1166 w, 1126 w, 1090 w, 1065 w, 1026 w, 958 w, 911 w, 827 w, 802 w, 775 w, 640 w, 618 w, 492 w.
Synthesis of μ-aqua-k2O:O-di-μ-2-methylpropionato-k4O:O'-bis[(2-methylpropionato-kO)bis(pyridi-ne-kN)nickel(II)] dihydrate[Ni2(μ-OH2)(μ-O2CCH(CH3)2)2(Рy)4((CH3)2CHCO2)2] ∙ 2H2O (II). 2‑Methylpropionic acid (0.75 mL, 8 mmol) was added to a suspension of NiCO3 ∙ 5.5H2O (0.8711 g, 4 mmol) in an acetonitrile–water (100 : 1) mixture (50 mL), and a solution of Рy (0.64 mL, 8 mmol) in acetonitrile (10 mL) was poured to the reaction mixture. Blue crystals suitable for X-ray structure analysis precipitated from the mother liquor during slow evaporation in air. The yield of complex II was ~85%.
For C36H54N4O11Ni2 | |||
Anal. calcd., % | C, 51.78 | H, 6.52 | N, 6.71 |
Found, % | C, 51.93 | H, 6.22 | N, 6.82 |
IR (ν, cm–1): 3436 w, 3075 w, 2964 w, 2924 w, 2866 w, 2325 w, 2102 w, 2051 vw, 1613 s, 1601 s, 1572 m, 1533 m, 1485 m, 1471 m, 1444 m, 1415 s, 1365 m, 1310 w, 1286 m, 1217 m, 1169 w, 1146 w, 1111 w, 1091 m, 1074 m, 1070 w, 1038 m, 1011 w, 919 w, 831 m, 759 m, 699 s, 651 m, 629 m, 566 m, 433 m.
Synthesis of μ-aqua-k2O:O-di-μ-2-methylpropionato-k4O:O')-bis[(2-methylpropionato-kO)(2,2'-bipy-ridine-k2N,N')nickel(II)][Ni2(μ-OH2)(μ-O2CCH-(CH3)2)2(Вipy)2((CH3)2-CHCO2)2] (III). 2‑Methylpropionic acid (1.12 mL, 12 mmol) was added to a suspension of NiCO3 ∙ 5.5H2O (1.3068 g, 6 mmol) in an acetonitrile–water (100 : 1) mixture (40 mL), and a solution of Вipy (0.9364 g, 6 mmol) in acetonitrile (10 mL) was poured to the reaction mixture. Blue crystals suitable for X-ray structure analysis precipitated from the mother liquor during slow evaporation in air. The yield of complex III was ~80%.
For C36H46N4O9Ni2 | |||
Anal. calcd., % | C, 54.39 | H, 5.84 | N, 7.02 |
Found, % | C, 54.50 | H, 5.52 | N, 7.23 |
IR (ν, cm–1): 3114 w, 3037 w, 2961 w, 2921 w, 2866 w, 2066 w, 1979 w, 1899 w, 1612 s, 1605 s, 1569 m, 1531 w, 1495 w, 1478 m, 1469 m, 1442 m, 1414 s, 1366 m, 1357 m, 1322 m, 1308 w, 1285 m, 1250 w, 1168 w, 1151 m, 1113 w, 1088 m, 1058 m, 1026 m, 1018 w, 919 w, 829 m, 763 s, 736 s, 652 m, 632 m, 553 m, 448 w, 417 m.
X-ray structure analyses were carried out on an Xcalibur diffractometer (Rigaku Oxford Diffraction) equipped with an Eos CCD detector (MoKα radiation, λ = 0.71073 Å, graphite monochromator) at 100(2) K for complex I and on an XtaLab Supernova diffractometer (Rigaku Oxford Diffraction) equipped with a HyPix-3000 detector (CuKα radiation, λ = 1.54184 Å, mirror monochromator) at 100(2) K for complexes II and III. The structures of the compounds were solved by direct methods and refined using the SHELX programs [33] integrated in the OLEX2 complex [34]. The final models included the coordinates and anisotropic thermal parameters for all non-hydrogen atoms. The positions of the hydrogen atoms of the organic fragments of the molecules were calculated using the algorithm implemented in the SHELX program. The positions of the hydrogen atoms of the organic molecules were calculated using the algorithm implemented in the SHELX program package, where Uiso(H) was established as 1.5Ueq(C) and C–H was 0.96 Å for the CH3 groups, Uiso(H) was established as 1.2Ueq(C) and C–H was 0.97 Å for the CH2 groups, Uiso(H) was established as 1.2Ueq(C) and C–H was 0.93 Å for the CH groups of the cyclic fragments, and Uiso(H) was established as 1.2Ueq(C) and C–H was 0.98 Å for the tertiary CH group. The positions of the hydrogen atoms of the H2O molecules were localized from the difference Fourier synthesis and fixed during refinement with 1.5Ueq(O). Note that the monodentate 2-methylpropionate ligands in the structure of complex II were disordered over two crystallographically nonequivalent positions with a general population of 1.0. The crystallographic data and experimental and structure refinement parameters for complexes I–III are presented in Table 1.
The crystallographic data for complexes I–III were deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 1840763 (I), 1913469 (II), and 1913471 (III); www.ccdc.cam.ac.uk/structures/).
RESULTS AND DISCUSSION
Synthetic hellyerite NiCO3 ∙ 5.5H2O was used as the initial compound in this work. The application of this compound allowed the fast syntheses in high yields of three new 2-methylpropionate complexes [Ni2(μ-OH2)(μ-O2CCH(CH3)2)2L2–4((CH3)2CHCO2)2] (L is Tmeda (I), Рy (II), and Bipy (III)).
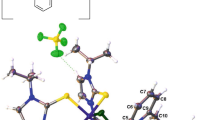
The molecular structures of complexes I–III are similar and have the binuclear framework in which two nickel atoms are linked to each other by the water molecule and two bidentate-bridging carboxylate ligands (Fig. 1). The coordination environment of each nickel ion is complemented by the 2-methylpropionate ligand coordinated via the monodentate mode and one bidentate N-donor ligand (Tmeda for I and Вipy for III) or two monodentate N-donor ligands (Рy for II). The structures of complexes I–III are stabilized by intramolecular hydrogen bonds between the bridging water molecule and carboxylate ligands coordinated via the monodentate mode. The lengths of the corresponding hydrogen bonds are presented in Table 2.

Molecular structure of complex I. Intramolecular hydrogen bonds are shown. Thermal ellipsoids are given with 50% probability.
In the cases of complexes I and III containing the chelating bidentate ligands Tmeda and Вipy, a trapezoidal distortion of the equatorial plane of the coordination polyhedron is observed (Fig. 2). The concept on the so-called “continuous symmetry measures” S(Oh) was used for the quantitative characterization of distortions of coordination polyhedra [35–37]. The value of continuous symmetry measures describes a deviation from the ideal octahedron shape: the higher the S(Oh), the more appreciable the deviations. The calculation of S(Oh) was performed by the X-ray structure data using the described algorithm [38]. For complexes I, II, and III, S(Oh) was 0.2044, 0.0744, and 0.6117, respectively, which indicates that the most significant deviations are observed for complex III, whereas complex II is characterized by minimum deviations. This can be related to a higher structural rigidity of the Вipy ligand in the complex compared to that of Tmeda. In the case of the Рy ligand, it cannot be excluded that its monodentate coordination mode assuming a higher flexibility of the metallic framework results in the lowest distortion of the coordination polyhedra.

Equatorial planes of the coordination polyhedra of complexes I–III. Carboxylate ligands and hydrogen atoms are omitted.
The π–π-stacking interactions of the aromatic ligands (Рy and Вipy) leading to the formation of 1D polymer chains with average interplanar distances of 3.282 and 3.363 Å are observed in the crystal structures of complexes II and III, respectively (Fig. 3). The crystal structure of complex II, in addition to π−π interactions, contains a system of hydrogen bonds between monodentate coordinated carboxylate ligands and solvate water molecules.

Fragments of the crystal structures of complexes (a) II and (b) III with the stacking interactions. The coordination polyhedra NiN2O4 are shown.
In the IR spectrum of complex II, the weak and broad peak at 3436 cm–1 indicates the presence of water of crystallization (νas(O–H) and νs(O–H)). The absorption bands in a range of 3100–2800 cm–1 correspond to the ν(C–H) vibrations of the methylene and methyl groups of Tmeda (for I) and radicals of the carboxylate ligands (I–III). The intense absorption bands νas(COO–) (1618 and 1616 for I, 1613 and 1601 for II, 1612 and 1605 cm–1 for III) and the νs(COO–) bands (1461 and 1414 for I, 1444 and 1415 for II, 1442 and 1414 cm–1 for III) belong to the bridging and monodentate carboxylate ligands, respectively [39].
The nickel atoms exist in the distorted octahedral environment of NiN2O4 in complexes I–III. Three spin-allowed transitions 3A2g → 3T2g, 3A2g → 3T1g, and 3A2g → 3T1g(P) [40, 41] at 7000–13 000, 11 000–20 000, and 19 000–27 000 cm–1, respectively, appear in the visible range for similar octahedral and pseudo-octahedral d8 systems. For complexes I–III, these transitions are observed at ~9500, ~15 500, and ~26 000 cm–1, respectively (Fig. 4, Table 3). In the case of complex III, a significant increase of the molar extinction coefficient of the 3A2g → 3T1g(P) transition can be explained by the intraligand charge transfer in the developed aromatic Вipy–ligand system. The shoulder at ~13 000 cm–1 (Fig. 4) can be explained by the low-intensity spin-forbidden transition 3A2g → 1Eg [40] observed at 12 630(11), 12 900(9), and 12 750(7) cm–1 (L cm–1 mol–1) for complexes I–III, respectively.

Electronic absorption spectra of complexes I–III in acetonitrile solutions at room temperature.
The study of the thermal behavior of complexes I–III showed that the thermal destruction of the complexes with the mono- (II) and bidentate (I and III) N-donor ligands proceeded via different routes. Complex II begins to decompose from the removal of outer-sphere water of crystallization, and this process ceases at 85°С (Δmtheor = 4.31, Δmexp = 4.87%). Then the combined consecutive elimination of the bridging water molecule and two pyridine molecules (Δmtheor = 21.07, Δmexp = 21.52%) is observed and possibly accompanied by the formation of compounds with the framework of the Chinese lantern type [21, 42]. Then two consecutive decomposition stages occur at 210–310 and 310–360°С to form nickel(II) oxide (Δmtheor = 56.75, Δmexp = 58.33%) (Fig. 5).

TG curves for complexes I–III.
The TG curve of complex I exhibits a mass loss of 2.5% in a range of 100–120°С, which corresponds to the loss of the bridging water molecule. A sharp mass loss of 65.3% is observed in a range of 140–300°С. The total mass loss is 88.2% at the 20.9% (based on oxide) nickel content in the complex, indicating that the sublimation of the complex occurs in parallel to the thermal destruction processes.
The decomposition of complex III starts from the removal of the bridging water molecule in a range of 100–130°С. The further heating results in a sharp mass loss of the sample (Δmexp = 65.27%) in a range of 230–370°С. Two stages of the decomposition of the complex corresponding to the final destruction of the coordination polyhedra with the formation of NiO (Δmtheor = 81.2, Δmexp = 80.5%) are observed in a range of 370–540°С.
To obtain information about possible forms of the complexes in acetonitrile solutions, we studied them by mass spectroscopy. Both the positive and negative detection ranges of the ESI-MS spectra of an acetonitrile solution of complex I exhibit low-intensity peaks corresponding to the molecular ions [M + H]+ (m/z = 714.2, 1%) and [M–H]– (m/z = 713.2, 1%). The ion corresponding to the mononuclear species [Ni(Тmeda)((CH3)2CHCOO)]+ (m/z = 261.1, 100%) has the highest intensity. Two peaks with m/z = 567.2 (17%) and 609.2 (12%) are of lower intensity. The second peak can be assigned to the [Ni2(O2CCH(CH3)2)3(Тmeda)2]+ ion.
A considerably higher number of peaks with high intensities corresponding to the “heavy” ions are observed in the mass spectra of complex II. The peaks with m/z = 799.2 (20%) and 781.8 (38%) were ascribed to the molecular ion [M + H]+ and its dehydrated form [M–H2O + H]+. In addition, two high-intensity peaks assigned to [Ni2(O2CCH(CH3)2)4(Рy)3 + H]+ (m/z = 702.2, 100%) and [Ni2(O2CCH(CH3)2)4(Рy)3 + MeCN + H]+ (m/z = 743.2, 75%) are observed. As in the case of complex I, the mononuclear species [Ni(O2CCH(CH3)2)2(Рy)2 + H]+ (m/z = 391.0, 36%) are detected in a solution of complex II. The dehydration of the complexes on the injection heated transfer line of the mass spectrometer can be one of the reasons for the absence of a water molecule in the structures of the most part of ions.
Thus, three new binuclear 2-methylpropionate complexes with the terminal N-donor ligands [Ni2(μ-OH2)(μ-O2CCH(CH3)2)2L2–4((CH3)2CHCO2)2] (L is Тmeda, Рy, and Вipy) were synthesized. The observed deviations of the obtained compounds from the ideal octahedron shape were calculated on the basis of the X-ray structure analysis data. The structurally more rigid chelating bidentate ligands Вipy and Тmeda induce the highest trapezoidal distortions of the equatorial plane of the coordination polyhedra (I < III), whereas Py coordinated via the monodentate mode assumes a higher flexibility of the metallic core of complex II leading to the lowest distortion. The π–π-stacking interactions resulting in the formation of 1D polymer chains are observed in the crystal structures of the complexes with the aromatic Py and Bipy ligands.
The thermal destruction of complex I is accompanied by the sublimation of the compound. In the case of complexes II and III, NiO is the final decomposition product. Complex II loses the Py ligands in a range of 120–200°С and is isomerized to form the structures of the Chinese lantern type.
According to the ESI-MS spectra, the partial elimination of the carboxylate ligands with the formation of the [Ni2(O2CCH(CH3)2)3(Tmeda)2]+ ions followed by their fragmentation to the mononuclear species [Ni(Тmeda)((CH3)2CHCOO)]+ predominates in acetonitrile solutions of complex I. One Tmeda ligand is retained in the coordination environment of nickel in all identified ions. In the case of complex II, the main process is the elimination of one Py ligand to form the binuclear complex cations [Ni2(O2CCH(CH3)2)4(Рy)3 + H]+ and [Ni2(O2CCH(CH3)2)4(Рy)3 + MeCN + H]+ undergoing the consequent fragmentation to a lower extent than complex I. The absence of the bridging water molecule in the structures of the most part of ions can be explained by the loss of this molecule under the electrospray conditions.
REFERENCES
Long, J.M., Gao, H.Y., Liu, F.Sh., et al., Inorg. Chim. Acta., 2009, vol. 362, no. 9, p. 3035.
Wang, J.-W., Liu, W.-J., Zhong, D.-Ch., and Lu, T.-B., Coord. Chem. Rev., 2019, vol. 378, p. 237.
Fischer, A.I., Panina, N.S., and Belyaev, A.N., Russ. J. Coord. Chem., 2016, vol. 42, no. 10, p. 635. https://doi.org/10.1134/S1070328416100018
Zorina-Tikhonova, E.N., Gogoleva, N.V., and Sidorov, A.A., Polyhedron, 2017, vol. 130, p. 67.
Malkov, A.E., Fomina, I.G., Sidorov, A.A., et al., J. Mol. Struct., 2003, vol. 656, nos. 1–3, p. 207.
Novotortsev, V.M., Rakutin, Yu.V., Nefedov, S.E., and Eremenko, I.L., Russ. Chem. Bull.,Int. Ed., 2000, vol. 49, no. 3, p. 438.
Nikolaevskii, S.A., Kiskin, M.A., Starikova, A.A., et al., Russ. Chem. Bull.,Int. Ed., 2016, vol. 65, no. 12, p. 2812.
Das, M., Harms, K., Ghosh, B.N., et al., Polyhedron, 2015, vol. 87, p. 286.
Matelková, K., Boča, R., Dlhán, L., et al., Polyhedron, 2015, vol. 95, p. 45.
Bottari, B., Maccari, R., Monforte, F., et al., Bioorg. Med. Chem., 2001, vol. 9, no. 8, p. 2203.
Bharty, M.K., Paswan, S., Dani, R.K., et al., J. Mol. Struct., 2017, vol. 1130, p. 181.
Rispens, M.T., Gelling, O.J., de Vries, A.H.M., et al, Tetrahedron, 1996, vol. 52, no. 10, p. 3521.
Dey, S.K., Salah, El., Fallah, M., Ribas, J., et al., Inorg. Chim. Acta, 2004, vol. 357, no. 5, p. 1517.
Chang, C.-H., Tsai, C.-Y., Lin, W.-J., et al., Polymer, 2018, vol. 141, p. 1.
Sumner, J.B., J. Biol. Chem., 1926, vol. 69, p. 435.
Blakeley, R.L., Treston, A., Andrews, R.K., and Zerner, B., J. Am. Chem. Soc., 1982, vol. 104, no. 2, p. 612.
Blakeley, R.L. and Zerner, B., J. Mol. Catal., 1984, vol. 23, nos. 2–3, p. 263.
Lippard, S.J., Science, 1995, vol. 268, no. 5213, p. 996.
Groom, C.R., Bruno, I.J., Lightfoot, M.P., and Ward, S.C., Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, vol. 72, p. 171.
Eremenko, I.L., Golubnichaya, M.A., Nefedov, S.E., et al., Russ. Chem. Bull.,Int. Ed., 1998, vol. 47, no. 4, p. 704.
Eremenko, I.L., Nefedov, S.E., Sidorov, A.A., et al., Inorg. Chem., 1999, vol. 38, no. 17, p. 3764.
Fomina, I.G., Sidorov, A.A., Aleksandrov, G.G., et al., Russ. Chem. Bull.,Int. Ed., 2004, vol. 53, no. 1, p. 114.
Denisova, T.O., Aleksandrov, G.G., Fialkovskii, O.P., and Nefedov, S.E., Zh. Neorg. Khim., 2003, vol. 48, no. 9, p. 1476.
Mikhailova, T.B., Fomina, I.G., Sidorov, A.A., et al., Zh. Neorg. Khim., 2003, vol. 48, no. 10, p. 1648.
Margossian, T. Larmier, K., Kim, S.M., et al., J. Am. Chem. Soc., 2017, vol. 139, no. 20, p. 6919.
Karmakar, A., Deka, K., Sarma, R.J., and Baruah, J.B., Inorg. Chem. Commun., 2006, vol. 9, no. 8, p. 836.
Karmakar, A., Sarma, R.J., and Baruah, J.B., Eur. J. Inorg. Chem., 2006, vol. 2006, no. 22, p. 4673.
Song, W.-D., Yan, J.-B., and Hao, X.-M., Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, vol. 64.
Singh, S., Saini, D., Mehta, S.K., and Choquesillo-Lasarte, D., J. Coord. Chem., 2011, vol. 64, no. 9, p. 1544.
Qadir, A.M., Asian J. Chem., 2013, vol. 25, no. 15, p. 8829.
Walsh, J.P.S., Sproules, S., Chilton, N.F., et al., Inorg. Chem., 2014, vol. 53, no. 16, p. 8464.
Bette, S., Rincke, C., Dinnebier, R.E., and Voigt, W., Z. Anorg. Allg. Chem., 2016, vol. 642, nos. 9−10, p. 652.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Alvarez, S., Avnir, D., Llunell, M., and Pinsky, M., New J. Chem., 2002, vol. 26, p. 996.
Alvarez, S., Chem. Rev., 2015, vol. 115, no. 24, p. 13447.
Pavlov, A.A., Nikovskii, I.A., Polezhaev, A.V., et al., Russ. J. Coord. Chem., 2019, vol. 45, p. 402. https://doi.org/10.1134/S1070328419060046
The Continuous Symmetry Group. http://www.csm.huji.ac.il/new/.
Nakamoto, K., Infrared and Raman Spectra of Inorganic and Coordination Compounds, Pt B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, New York: Wiley, 2009.
Lever, A.B.P., Inorganic Electronic Spectroscopy, Amsterdam: Elsevier, 1984.
Meredith, P.L. and Palmer, R.A., Inorg. Chem., 1971, vol. 10, no. 5, p. 1049.
Lee, D., Hung, P.-L., Spingler, B., and Lippard, S.J., Inorg. Chem., 2002, vol. 41, no. 3, p. 521.
ACKNOWLEDGMENTS
The structural studies were carried out at the Resource Center “X-ray Diffraction Investigation Methods” of the Scientific Park of the St. Petersburg State University. The IR spectra were recorded and thermogravimetry was conducted at the Engineering Center of the St. Petersburg State Institute of Technology (Technical University).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Blinou, D.O., Nikiforov, A.A., Gurzhiy, V.V. et al. Complexes [Ni2(μ-OH2)(μ-O2CCH(CH3)2)2L2–4((CH3)2CHCO2)2]: Synthesis, Structure, and Mass Spectrometric Studies. Russ J Coord Chem 46, 81–88 (2020). https://doi.org/10.1134/S1070328420020037
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328420020037