
Abstract
The reactions of the complex [Co(DAP)2(Piv)2] · Piv (Ia), where Piv is the pivalate anion, DAP is 1,3-diaminopropane, with Cd(II) and Li(I) pivalates afford heterometallic compounds, molecular complex [CdCo(DAP)2(Piv)5] (I) and polymeric complex [LiCo(DAP)2(Piv)4]n (II) (CIF files CCDC nos. 1908223 and 1908224, respectively). According to X-ray photoelectron spectroscopy, the electronic structure of cobalt ions in complexes Ia and I corresponds to low-spin Co(III) surrounded by strong-field ligands. The thermal destruction of I and II, studied by synchronous thermal analysis, includes stages of desorption of solvate molecules, destruction of the 1,3-diaminopropane molecule with nitrogen evolution, and degradation of carboxylate anions. The final product of thermolysis of I is the complex oxide CdCoO2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Numerous data on the synthesis and properties of stable molecular heterometallic carboxylate complexes [1–11] and the fact that the initial metal core is retained upon the synthesis of framework coordination polymers from these complexes [12–14] indicates good prospects of the search and study of new heterometallic systems. Cobalt(III) carboxylate complexes, which are readily synthesized by the reaction of cobalt(II) dipivalate with primary amines in the presence of atmospheric oxygen [15], attract attention as the starting species for such studies. Unlike Fe(III) or Mn(III) carboxylate complexes [10, 16, 17], Co(III) complexes devoid of bridging O2– anions can be prepared rather easily; this, in turn, implies the possibility to obtain heterometallic compounds using the same procedures as used to prepare complexes of 3d metals in +2 oxidation state.
The purpose of this study is to establish the composition and structure of the products of reaction of the previously obtained [Co(DAP)2(Piv)2] · Piv (Ia) complex (where Piv is the pivalate anion, DAP is 1,3-diaminopropane) [15] with Cd(II) and Li(I) pivalates. Cadmium was chosen because of the large ionic radius of Cd(II), which allows the metal center to coordinate more donor atoms (up to 8) than is coordinated by 3d metal ions and, hence, the formation of a heterometallic compound is more likely in this case. The difference between the metal ionic radii also simplifies the X-ray diffraction identification of atoms. Prospects for the formation of Li–Co complexes are due to the existence of stable tetranuclear [Li2M2(Piv)6L2] compounds (M = Co, Zn) [12, 18–20] and metal-organic framework polymers (MOFP) based on them [12, 13, 20]. The search for the starting complexes for the synthesis of Li(I)–Co(III) MOFPs is a relevant task for expanding the range of frameworks of new type with definite functional properties.
EXPERIMENTAL
The complexes were prepared using commercial chemicals and solvents without further purification: pivalic acid (HPiv) (Merck), CoCl2 · 6H2O (Khim-med, analytical grade), Cd(NO3)2 · 4H2O (Acros), benzene (analytical grade), acetonitrile (Khimmed, special purity grade), DAP (Acros). Lithium pivalate Li(Piv) was prepared by the reaction of equimolar amounts of aqueous solutions of LiOH and HPiv followed by evaporation; the resulting solid phase was washed with hexane. Complex Ia was synthesized by the procedure proposed in [15]. Anaerobic thermolysis was conducted in an argon flow (Ar, >99.998%; O2, 0.0002%; N2, <0.001%; H2O, <0.0003%; CH4, <0.0001%). The starting polymeric complex [Co(OH)n(O)(Piv)2 – n]x was synthesized by a reported procedure [21] and characterized by IR spectroscopy and chemical analysis data.
IR spectra were recorded on a Perkin-Elmer Spectrum 65 Fourier transform IR spectrophotometer in the attenuated total reflection (ATR) mode in the 400–4000 cm–1 frequency range.
Elemental analysis was performed on an automated Carlo Erba EA 1108 C,H,N,S-analyzer.
X-ray photoelectron spectra (XPS) were measured on a Kratos Axis Ultra spectrometer (monochromic AlKα radiation with a power not exceeding 180 W). The surface charge of the samples was neutralized using a low-energy electron gun. The spectra were resolved into components using the Kratos Analytical program. Each spectral line was approximated by a Gaussian profile or their sum. The measurements were repeated at least twice at a pressure of ~10–9 Torr. The spectra were measured at both liquid nitrogen and room temperatures. The spectra were calibrated against the C1s line; the binding energy of the component corresponding to the C–(C,H) bond was taken to be 285.0 eV. The results were in agreement with X-ray diffraction data.
Synthesis of [CdCo(DAP)2(Piv)5] ∙ 1/2C6H6 (I). CdPiv2 (0.3 g, 0.7 mmol) and a solution of 1,3-diaminopropane (0.13 mL, 1.5 mmol) in benzene (10 mL) were added to a suspension of [Co(OH)n(O)(Piv)2 – n]x (0.2 g, 0.8 mmol) in acetonitrile (50 mL). The resulting cherry-colored solution was heated (70°C) for 40 min, filtered, and concentrated. The claret-colored crystals of I that precipitated within 24 h were separated from the mother liquor by decantation, washed with cold diethyl ether, and dried in an argon flow. The yield of I was 120 mg (60% for the desolvated product dried to a constant weight).
For C34H68N4O10CoCd (I) | |||
Anal. calcd., % | C, 45.21 | H, 7.59 | N, 6.20 |
Found, % | C, 44.89 | H, 7.61 | N, 6.43 |
IR (ν, cm–1): 3262 br.m, 3224 br.m, 3154 w, 2960 s, 2930 m, 2870 m, 2300 vw, 2253 w, 1620 m, 1583 w, 1540 vs, 1480 vs, 1458 w, 1410 vs, 1356 vs, 1336 vs, 1306 w, 1211 br.s, 1136 w, 1112 m, 1050 br.m, 924 m, 898 vs, 807 m, 792 m, 752 br.w, 682 w, 640 w, 604 m, 571 w, 521 m, 460 m, 425 w.
Synthesis of [LiCo(DAP)2(Piv)4]n (II). LiPiv (0.08 g, 0.7 mmol) and a solution of 1,3-diaminopropane (0.13 mL, 1.5 mmol) in benzene (10 mL) was added to a suspension of [Co(OH)n(O)(Piv)2 – n]x (0.2 g, 0.8 mmol) in acetonitrile (50 mL). The resulting pink suspension was heated (70°C) for 40 min. The pink crystals of II that precipitated within 24 h were separated from the mother liquor by decantation, washed with cold diethyl ether, and dried in an argon flow. The yield of II was 70 mg (35% for the desolvated product dried to a constant weight).
For C29H59N4O8LiCo (II) | |||
Anal. calcd., % | C, 49.99 | H, 8.54 | N, 8.04 |
Found, % | C, 50.18 | H, 8.38 | N, 7.99 |
IR (ν, cm–1): 3297 w, 3200 br.w, 3153 w, 3089 w, 2953 m, 2930 m, 2866 m, 1638 m, 1595 vs, 1564 vs, 1479 vs, 1454 w, 1421 w, 1346 vs, 1302 m, 1261 w, 1200 br.w, 1111 m, 1047 m, 1036 m, 931 m, 907 w, 895 w, 881 m, 789 m, 726 br.w, 679 vs, 648 m, 592 m, 577 w, 547 w, 528 w, 446 vs, 423 w.
X-ray diffraction analysis of the crystals of I and II was performed on Bruker Apex II and Bruker Apex II DUO diffractometers equipped with CCD detectors (MoKα-radiation, λ = 0.71073 Å, graphite monochromator) at 120 K. The structures were solved by the direct method and refined by full-matrix least squares method in the anisotropic approximation using SHELXL-2014/7 [22] and Olex2 [23] software. The hydrogen atoms of the NH2 groups were located from difference Fourier maps, the positions of other hydrogen atoms were calculated geometrically. All hydrogen atoms were refined isotropically in the riding model. The crystallographic parameters and refinement details of complexes I and II are given in Table 1.
The full set of X-ray diffraction data is deposited with the Cambridge Crystallographic Data Centre (CCDC nos. 1908223 (I), 1908224 (II); deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac. uk/data_request/cif).
Powder X-ray diffraction analysis of both samples was carried out on a Bruker D8 Advance instrument (CuKα, λ = 1.54 Å, Ni-filter, LYNXEYE-detector).
The thermal behavior of I and II was studied by synchronous thermal analysis (STA) under argon with simultaneous recording of thermogravimetric (TG) and differential scanning calorimetric (DSC) curves. The measurements were carried out on an STA 449 F1 Jupiter (NETZSCH) in aluminum crucibles covered by lids with holes to maintain 1 atm pressure during the thermal decomposition. The samples were heated to 450°C at a rate of 10°C/min. The sample mass was 5.57 mg (I)/1.13 mg (II). The accuracy of temperature measurements was ±0.7°C and the accuracy of mass change measurement was ±1 × 10−2 mg. The TG and DSC curves were recorded using the correction file and temperature and sensitivity calibrations for the specified temperature program and heating rate. After thermal analysis, qualitative chemical composition and micromorphology of the residue were determined using a Carl Zeiss NVision 40 scanning electron microscope equipped with an Oxford X-Max X-ray spectroscopic detector at accelerating voltage of 1 and 20 kV, respectively. The magnification ranged from ×3000 to ×300 000.
RESULTS AND DISCUSSION
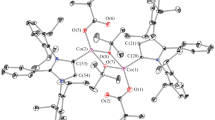
The reaction of [Co(DAP)2(Piv)2] · Piv (Ia) with cadmium dipivalate [24] in benzene affords molecular binuclear heterometallic complex [CdCo(DAP)2-(Piv)5] or [CdII(κ2-Piv)3(µ2-Piv)CoIII(κ2-DAP)2(κ1-Piv)] (I, Fig. 1). According to X-ray diffraction data for its 1 : 0.5 benzene solvate, the cobalt(III) ion has the same distorted octahedral environment as found in the starting compound Ia. It is formed by two chelating propanediamine molecules and two pivalate anions (with similar Co–N and Co–O(4) distances, Table 2), one of which occupies a bridging position between the Co3+ and Cd2+ ions. The outer-sphere pivalate anion of the starting complex Ia completes the Cd(II) coordination environment, being chelated to it to give {Cd(κ2-OOC–)3(κ1-OOC–)}, and forms a distorted one-cap trigonal prism around cadmium. In this case, two of the four NH2 groups of the propanediamine ligands form intramolecular hydrogen bonds with terminal, bridging, and two chelating pivalate anions (N···O, 2.848(4)–3.057(5) Å; NHO, 123.0(4)°–152.5(3)°). The other two NH2 groups are connected to pivalate anions coordinated to Cd(II) in the neighboring complex I molecule (N···O, 2.874(5)–2.964(7) Å; NHO, 139.5(6)°–156.9(3)°) to give a hydrogen-bonded infinite chain.

Structure of [CdCo(DAP)2Piv5] (I) with atoms drawn as thermal ellipsoids (p = 50%). Hydrogen atoms, except those of the NH2 groups of the ligands, and benzene solvate molecule are omitted for clarity; tert-Bu stand for the tert-butyl groups of the pivalate anions.
Since upon the formation of I, one Piv– anion that is bonded to Co(III) in the monodentate fashion in the starting compound switches to the bridging position, an attempt was made to obtain the heterometallic coordination polymer [Cd2Co(DAP)2Piv7] by using two equivalents of [Cd(H2O)2(Piv)2] in the reaction. However, no heterometallic compounds besides I were isolated from the solution.
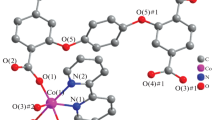
The reaction of Ia with LiPiv results in the formation of 1D coordination polymer [LiCo(DAP)2-(Piv)4]n or [Li(κ1-Piv)2(µ2-Piv)2Co(DAP)2]n (II, Fig. 2). The Co(III) coordination environment also remains unchanged (Table 2), but both Piv– anions, which are coordinated to cobalt(III) in the monodentate fashion in the starting compound, occupy bridging positions between cobalt and Li+. This binding gives rise to an infinite 1D chain in which the alternating lithium and cobalt ions are linked by a bridging carboxylate anion. In turn, the lithium(I) ion is surrounded by two more pivalate anions, which form a distorted tetrahedral environment, most typical of Li+. The lithium-uncoordinated oxygen atoms are hydrogen-bonded to the diaminopropane NH2 groups (N…O, 2.848(2)–3.027(3) Å; NHO, 148.1(2)o–175.5(3)o); this additionally stabilizes the resulting 1D-coordination polymer. In the crystal, these chains are connected only by weak contacts rather than by hydrogen bonds involving the N(2)H2 group; the formation of hydrogen bonds is apparently precluded by shielding of the corresponding hydrogen atoms by bulky tert-butyl substituents of Piv–.

Structure of 1D-coordination polymer [LiCo(DAP)2(Piv)4]n (II) with atoms drawn as thermal ellipsoids (p = 50%). Hydrogen atoms, except those of the NH2 groups of the ligand, and benzene solvate molecule are omitted for clarity; tert-Bu stand for the tert-butyl groups of the pivalate anions. The cobalt(III) ion occupies a special position (inversion center) in the crys-tal.
The oxidation state of cobalt in the starting compound Ia and heterometallic complex I was confirmed by XPS. Most often, transition from lower to higher oxidation state results in a higher binding energy (Eb) of the ground-state level. However, in some cobalt compounds, high-spin Co(II) states have higher Eb values than the low-spin Co(III) states [25]. In the case of cobalt, the Co2p spin–orbit splitting (ΔE) is more informative [25, 26]. The ΔE value is the binding energy (Eb) difference between the Co2p1/2 and Co2p3/2 lines. The results are summarized in Table 3.
The Co2p XPS spectra for samples I and Ia recorded at liquid nitrogen temperature (–77 K) are shown in Fig. 3a.

Co2p X-ray photoelectron spectra: (a) I and II (–77 K); (b) I (273 K): (a) beginning and (b) end of the experiment.
The Co2p3/2 spectrum of these complexes is a rather narrow line with a weak satellite component and with ΔE of 14.9 and 14.8 eV (Table 3), which corresponds to Co(III). According to theoretical calculations [27], the spin–orbit splitting increases depending on the number of unpaired 3d electrons. Previous experiments [25, 26, 28] confirm the theoretical result: this splitting for diamagnetic Co(III) compounds and paramagnetic Co(II) compounds is close to 15 and 16 eV, respectively. It is noteworthy that the first scans of Ia at room temperature detected a single form of cobalt in +3 oxidation state, with the spectrum being fully identical to the spectrum of complex I recorded at –77 K. By the end of the measurement, cobalt has been partially reduced to Co(II) under the action of X-rays (Fig. 3b).
The lack of stability of I to X-rays may be attributable to structural parameters of this complex. It is known that transfer to the low-spin state is accompanied by suppression of satellites in the 3d metal spectra, which attests to decreasing ionicity of the metal–ligand bond [29]. The Co2p XPS spectrum of complexes I and Ia (Fig. 3a) has a weak satellite component, indicating the diamagnetic state of cobalt. This means that the occupancy of 3d shell is 6. The only way to explain the absence of spin on the cobalt atoms in this configuration is to classify these compounds as complexes with strong-field ligands. In this case, six 3d electrons occupy the triply degenerate 3d(t2g) level, while the energy increase caused by antiparallel spin orientation is counterbalanced by a decrease in the t2g energy in the ligand field. It is known [30] that among 3d metals, Co(III) atoms are most likely to form diamagnetic compounds such as complexes with strong-field ligands. The assignment of oxygen O1s and nitrogen N1s lines (Table 3) corresponds to the structural formulas of the samples. Thus, relying on analysis of the Co2p XPS spectra, complexes Ia and I were identified as strong-field complexes containing Co(III) in the diamagnetic state.
The thermal behavior of complexes I and II was studied by STA (with simultaneous recording of TG and DSC curves) under argon up to 450°C. The different natures of heteroatoms incorporated in the complexes and different modes of structural organization (I is a molecular complex; II is a polymer) account for the crucially different thermal decomposition routes (Fig. 4). Complex I is stable up to 125°C. The following stages of thermolysis can be distinguished in the TG curve (Fig. 4a, Table 4). The first one takes place in the 125–206°C range and includes two minor endotherms in the DSC curve (Fig. 4b) with peaks at 167 and 196°C. Two close endotherms correspond to desorption of two DAP molecules with evolution of two N2 molecules (mtheor/exp = 6.2/6.3%).

(a) TG and (b) DSC curves for complexes (a) I and (b) II.
The degradation of the nitrogen-containing moiety is followed by degradation of {Cd(Piv)3} (the fact that the nitrogen-containing moiety is the first to degrade is indicated by Ediss of the bonds: N–C, 70; O–C, 84 kcal/mol [31]). In the DSC curve, this stage (Fig. 4b) gives rise to a highly intense exothermic peak (extremum at 258°C) corresponding to sublimation of gaseous products of thermolysis of the {Cd(Piv)3} group (mtheor/exp = 33.5/32.3%). (Thermolysis of the single [Cd(Piv)2]n complex (Table 4) also demonstrates that thermal decomposition is accompanied by highly intense exothermic peak with an extremum at 412°C.) The mass loss for the TG curves of I (Fig. 4a) in the range of 206–589°C corresponds to the gradual destruction of the pivalate moiety (mtheor/exp = 61.0/58.8%). This is consistent with the data from previous experiments for pivalate complexes [29–35]).
Complex II is thermally less stable than I and destruction processes already start at 89°C (Fig. 4b, Table 4). The first stage corresponds to degradation of diaminopropane with evolution of two nitrogen molecules (mtheor/exp = 8.04/8.07%). The temperature characteristics of this process in complex II are somewhat shifted to lower temperature compared with I (Table 4). The stage of mass loss (28.8%) clearly seen in TG curve imples the removal of two Piv– groups hydrogen-bonded to DAP molecules (mtheor = 28.9%) (Figs. 2, 4b). The final stage corresponds to destruction of the residual organic groups (the total mass loss is 79.6%; mtheor = 79.4%). The ultimate mass (8%) attests to volatility of the complex (after the experiment, metal traces were visible on the crucible lid). Unlike II, in the case of I, the residual weight amounts to 24.8%, which corresponds to CdCoO2 (mtheor = 22.5%; the somewhat greater mass is due to carbonization of the residue after thermolysis in an inert atmosphere). The highly intense carbon peak in the energy dispersive spectrum is attributable to the substrate material (Fig. 5).

(a) Energy dispersive spectrum and (b, c) micromorphology images (b, ×10000; c, ×30000) of the residue of I.
Thus, it was shown that mononuclear complex Ia reacts with cadmium and lithium pivalates to form binuclear heterometallic molecular complex I and heterometallic 1D coordination polymer II, respectively. The cobalt coordination environment and +3 oxidation state in these compounds are retained. The replacement of trimethylacetate anions by dicarboxylic (e.g., terephthalic) acid anions provides the real possibility to prepare new heterometallic framework polymers with Co(III) atoms. The retention of the initial coordination environment gives hope that it would also be retained in dicarboxylate (or polycarboxylate)-bridged frameworks.
REFERENCES
Sidorov, A.A., Fomina, I.G., Talismanov, S.S., et al., Russ. J. Coord. Chem., 2001, vol. 27, no. 8, p. 548.
Mikhailova, T.B., Fomina, I.G., Sidorov, A.A., et al., Zh. Neorg. Khim., 2003, vol. 48, no. 10, p. 1648.
Malkov, A.E., Fomina, I.G., Sidorov, A.A., et al., Izv. Akad. Nauk. Ser. Khim., 2003, no. 2, p. 489.
Mikhailova, T.V., Pakhmutova, E.V., Malkov, A.E., et al., Izv. Akad. Nauk. Ser. Khim., 2003, no. 10, p. 1994.
Pakhmutova, E.V., Malkov, A.E., and Mikhailova, T.V., Izv. Akad. Nauk. Ser. Khim., 2003, no. 1, p. 131.
Malkov, A.E., Fomina, I.G., Sidorov, A.A., et al., J. Mol. Struct., 2003, vol. 656, nos. 1−3, p. 207.
Fomina, I.G., Sidorov, A.A., Aleksandrov, G.G., et al., Izv. Akad. Nauk. Ser. Khim., 2004, no. 7, p. 1422.
Aleksandrov, G.G., Fomina, I.G., Sidorov, A.A., et al., Izv. Akad. Nauk. Ser. Khim., 2004, no. 6, p. 1153.
Sidorov, A.A., Nikifirova, M.E., Pakhmutova, E.V., et al., Izv. Akad. Nauk. Ser. Khim., 2006, no. 11, p. 1851.
Kiskin, M.A., Fomina, I.G., Aleksandrov, G.G., et al., Inorg. Chem. Commun., 2004, vol. 7, no. 6, p. 734.
Eremenko, I.L., Kiskin, M.A., Fomina, I.G., et al., J. Cluster Sci., 2005, vol. 16, no. 3, p. 331.
Sapianik, A.A., Zorina-Tikhonova, E.N., Kiskin, M.A., et al., Inorg. Chem., 2017, vol. 56, p. 1599.
Sapianik, A.A., Kiskin, M.A., Samsonenko, D.G., et al., Polyhedron, 2018, vol. 145, p. 147.
Sap’yanik, A.A., Lutsenko, I.A., Kiskin, M.A., et al., Izv. Akad. Nauk. Ser. Khim., 2016, no. 11, p. 2106.
Sidorov, A.A., Talismanova, M.O., Fomina, I.G., et al., Izv. Akad. Nauk. Ser. Khim., 2001, no.. 11, p. 2106.
Kiskin, M.A., Sidorov, A.A., Fomina, I.G., et al., Inorg. Chem. Commun., 2005, vol. 8, no. 6, p. 524.
Kiskin, M.A., Fomina, I.G., Aleksandrov, G.G., et al., Inorg. Chem. Commun., 2005, vol. 8, no. 1, p. 89.
Dobrokhotova, Z., Emelina, A., Sidorov, A., et al., Polyhedron, 2011, vol. 30, p. 132.
Dobrohotova, Z.V., Sidorov, A.A., Kiskin, M.A., et al., J. Solid State Chem., 2010, vol. 183, p. 2475.
Sapianik, A.A., Kiskin, M.A., Kovalenko, K.A., et al., Dalton Trans., 2019, vol. 48, p. 3676.
Fomina, I.G., Aleksandrov, G.G., Dobrokhotova, Zh.V., et al., Izv. Akad. Nauk. Ser. Khim., 2006, p. 1841.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, p. 112.
Bourhis, L.J., Dolomanov, O.V., Gildea-Bourhis, R.J., et al., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. 71, p. 59.
Gogoleva, N.V., Sidorov, A.A., Nelyubina, Yu.V., et al., J. Coord. Chem., 2018, vol. 44, no. 4, p. 219. https://doi.org/10.1134/S107032841808002X
Frost, D.C., McDowell, C.F., and Woolsey, I.S., Mol. Phys., 1974, vol. 27, p. 1473.
Okamoto, Y., Nakano, H., and Imanaka, T., Bull. Chem. Soc. Jpn., 1975, vol. 48, p. 1163.
Barinskii, R.L. and Nefedov, V.I., Rentgeno-spektral’noe opredelenie zaryada atomov v molekulakh (X-ray Spectroscopic Determination of Atomic Charges in Molecules), Moscow: Nauka, 1966.
Ivanova, T., Naumkin, A., Sidorov, A., et al., J. Electron Spectrosc. Relat. Phenom., 2007, vols. 156−158, p. 200.
Burger, K., Furlani, C., and Mattogno, G., Relat. Phenom., 1980, vol. 21, p. 249.
Skvortsova, L.I. Magnetometricheskii metod analiza (Magnetochemical Method of Analysis), Novosibirsk: Geo, 2006.
Lidin, R.A., Andreeva, L.L., and Molochko, V.A., Konstanty neorganicheskikh veshchestv (Parameters of Inorganic Compounds), Moscow: Drofa, 2008.
Lutsenko, I.A., Kiskin, I.A., Efimov, N.N., et al., Polyhedron, 2017, vol. 137, p. 165.
Lutsenko, I.A., Kiskin, M.A., Aleksandrov, G.G., et al., Izv. Akad. Nauk. Ser. Khim., 2018, no. 3, p. 449.
Lutsenko, I.A., Kiskin, M.A., Nelyubina, Y.V., et al., Polyhedron, 2019, vol. 159, p. 426.
Fomina, I.G., Aleksandrov, G.G., Dobrokhotova, Zh.V., et al., Izv. Akad. Nauk. Ser. Khim., 2006, no. 11, p. 1841.
ACKNOWLEDGMENTS
Single crystal and powder X-ray diffraction studies were carried out using the equipment of the Center for Molecular Structure Studies, Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences. Elemental analysis and IR spectroscopy measurements were done using the equipment of the Center for Collective Use of Physical Investigation Methods, Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences.
Funding
This work was supported by the Russian Foundation for Basic Research (project no. 18-29-04043).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by Z. Svitanko
Rights and permissions
About this article

Cite this article
Lutsenko, I.A., Nelyubina, Y.V., Primakov, P.V. et al. New Heterometallic Co(III) Pivalate Complexes with 1,3-(CH2)3(NH2)2. Russ J Coord Chem 45, 759–766 (2019). https://doi.org/10.1134/S1070328419110046
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328419110046