
Abstract
New lanthanide complexes with 4-(2,1,3-benzothiadiazol-4-ylamino)pent-3-en-2-onate (L–) [Ln(L)3] are synthesized using two methods: by the reaction of Ln(N(SiMe3)2)3 with the protonated form of the ligand LH (Ln = Sm, Gd) and by the reaction of LnCl3 (Ln = Eu) with LH in the presence of KN(SiMe3)2 as a base. According to the X-ray diffraction data, the synthesized complexes [Ln(L)3] · 0.5Solv (Solv = THF, C7H8) are isotypical (CIF files CCDC nos. 1826520, 1826521, and 1826522 for Sm, Eu, and Gd, respectively). A specific feature of the structures is the disordering of the metal atom and one of the ligands over two positions when the fragments of the ligands are arranged via the head-to-tail type occupying the same volume of the space. This probably leads to failure in the crystal lattice. The photoluminescence spectra of [Sm(L)3] · 0.5THF are recorded. A relationship between the coordination mode of the ligand and the position of the long-wavelength band of the electron transitions in the complexes with the L– ligand is revealed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
2,1,3-Benzothiadiazole (Btd) and its derivatives are of interest as precursors and/or components of materials for organic light emitting diodes and other optoelectronics: telecommunication systems, sensors, solar energy converters, and others [1, 2]. Among these objects, the most attention is presently given to organic polymers based on Btd characterized by a series of important properties necessary for the production of luminescent functional materials: (1) they exhibit electron-acceptor properties and can act as electron buffers [3–6], (2) they are efficient fluorophores [7–10], and (3) in the solid state they form supramolecular structures due to sulfur–nitrogen contacts and π–π interactions in the conjugated systems [11]. The coordination molecular complexes and coordination polymers of d and f elements with the functionalized Btd derivatives are of interest along with the conjugated polymer systems. They make it possible to eliminate some drawbacks of organic polymers, for example, to enhance the operating stability of light emitting diodes due to higher thermal, chemical, and photochemical resistance and to eliminate restrictions to quantum efficiency.
The nontrivial luminescence properties have been shown previously for some complexes with the Btd derivatives acting as ligands [12–17]. However, the compounds of both d and f metals with the Btd derivatives as ligands are still poorly studied and, hence, it is urgent to synthesize them, reveal their coordination modes, and study the properties, first of all, luminescence properties.
We have recently synthesized the Zn, Ag, and Cd complexes with 4-amino-2,1,3-benzothiadiazoles [18–21] and the lanthanide complexes with 4-oxy-2,1,3-benzothiadiazolate (O-Btd)− [22, 23] characterized by interesting photophysical properties. For example, (O-Btd)− was found to exhibit the properties of a ligand-antenna in the Er and Yb complexes: they are characterized by intense luminescence in the IR range [22, 23]. In addition, we synthesized a new representative of the class of functionalized benzothiadiazoles, 4-(2,1,3-benzothiadiazol-4-ylamino)pent-3-en-2-one (LH, Scheme 1), manifesting an intense emission and a high quantum yield [18]. The readily deprotonated ketoimine group makes LH to be a prospective chelating ligand toward d and f metals. We earlier reported the synthesis and study of the structure and properties of the [Zn(L)2] complex [18].
This work is aimed at developing the coordination chemistry of this ligand, i.e., elaborating the synthesis methods and X-ray diffraction analysis of the samarium, europium, and gadolinium complexes with L–.

Scheme 1 .
EXPERIMENTAL
Commercially available anhydrous LnCl3 (Ln = Sm, Eu, Gd) were used as received. Lanthanide amides Ln(N(SiMe3)2)3 (Ln = Sm, Gd) [24] and ketoimine LH [18] were synthesized using known procedures. The solvents were dehydrated by reflux and distillation over sodium under argon, degassed by multiple evacuating, and stored over a potassium–sodium alloy from where they were loaded to a reaction flask by condensation in vacuo. The complexes were synthesized and isolated in evacuated vessels without an inert gas. The samples for physicochemical analyses were prepared in an argon glove box to prevent hydrolysis. Elemental analyses for C, H, N, and S were carried out on a Euro EA 3000 instrument. Somewhat overestimated carbon and hydrogen contents in the [Gd(L)3] · 0.5C7H8 complex are related, most likely, to the incomplete removal of the solvent from the solid sample. The luminescence spectra of the solid samples were recorded on a Horiba Jobin Yvon Fluorolog 3 fluorescence spectrophotometer at 298 K (gap 5 × 5 nm, excitation wavelength 380 nm for LH and 450 nm for [Sm(L)3] · 0.5THF). The intensities of the radiation source and spectral response were corrected using standard correction curves. The spectra of the solid substances were recorded for powdered samples placed between two nonfluorescent quartz glasses.
X-ray diffraction analyses of compounds [Ln(L)3] · 0.5Solv were carried out on a Bruker APEX DUO diffractometer (МоКα radiation, λ = 0.71073 Å, graphite monochromator) at 150 K. The reflection intensities were measured using φ and ω scan modes for narrow (0.5°) frames. An absorption correction was applied using the SADABS program [25]. The structures were solved by a direct method and refined by full-matrix least squares in the anisotropic (for non-hydrogen atoms) approximation using the SHELXTL program package [26, 27] and OLEX2 graphical interface [28].
Hydrogen atoms were localized geometrically and refined by the riding model. The Ln atoms and one ligand are disordered over two positions with the general refined site occupancy from 92 : 8 to 84 : 16%. The C, N, and O atoms of the fragments with lower site occupancies were refined in the isotropic approximation with restraints to some bond lengths (DFIX), angular distances (DANG), and deviations of atoms from the plane of the C6 ring (FLAT). The disordering over two positions with a fixed site occupancy of 50 : 50% was refined for the atoms of the solvate THF and acetone molecules lying near the inversion center. The phenyl ring of toluene was refined as a rigid fragment (the DFIX restraint was imposed on the C–CH3 bond length). The corresponding restraints were also imposed on the bond lengths and angular distances of the THF molecules. The crystallographic data are presented in Table 1.
The X-ray diffraction data for the structures of [Ln(L)3] · 0.5Solv (Ln = Sm, Eu, Gd) were deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 1826520–1826522; deposit@ ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk/ data_request/cif) and are available from the authors.
Synthesis of [Sm(L)3] · 0.5THF. Tetrahydrofuran (∼10 mL) was condensed onto a mixture of solid LH (0.045 g, 0.193 mmol) and Sm(N(SiMe3)2)3 (0.040 g, 0.0633 mmol) cooled with liquid nitrogen. The obtained red solution was stirred for 24 h at room temperature and then concentrated to a volume of ∼2 mL. Red crystals of the product that formed were filtered off and dried in vacuo. The yield was 0.010 g (20%).
For C33H30N9O3S3Sm · 0.5C4H8O | ||||
Anal. calcd., % | C, 47.6 | H, 3.9 | N, 14.3 | S, 10.9 |
Found, % | C, 47.7 | H, 4.0 | N, 14.0 | S, 10.0 |
Synthesis of [Gd(L)3] · 0.5C7H8. Toluene (∼10 mL) was condensed onto a mixture of solid Gd(N(SiMe3)2)3 (0.050 g, 0.078 mmol) and LH (0.056 g, 0.24 mmol) cooled with liquid nitrogen. The reaction mixture was stirred at 50°С for 3 days. A dark red solution was filtered and evaporated to dryness. The obtained oily residue was washed with hexane to obtain a powdered product, which was dried in vacuo. The yield was 0.043 g (60%). The crystals suitable for X-ray diffraction analysis were obtained by the evaporation of a solution of the complex in a toluene–THF (1 : 1) mixture.
For C33H30N9O3S3Gd ∙ 0.5C7H8 | ||||
Anal. calcd., % | C, 48.7 | H, 3.8 | N, 14.0 | S, 10.7 |
Found, % | C, 49.8 | H, 4.6 | N, 13.6 | S, 10.6 |
Reaction of EuCl3 with LH and KN(SiMe3)2. Tetrahydrofuran (∼10 mL) was condensed onto a mixture of solid LH (0.068 g, 0.29 mmol), KN(SiMe3)2 (0.057 g, 0.028 mmol), and EuCl3 (0.026 g, 0.10 mmol) cooled with liquid nitrogen. The obtained red solution with a whitish precipitate was stirred at 50°C for 3 days. The solution was filtered and concentrated to 1 mL, which was accompanied by the formation of a minor amount of suitable for X-ray diffraction analysis red crystals of compound [Eu(L)3] · 0.5THF.
RESULTS AND DISCUSSION
The reaction of silylated amides Ln(N(SiMe3)2)3 with LH (Scheme 1) was chosen for the synthesis of the Sm(III) and Gd(III) complexes with 4-(2,1,3-benzothiadiazol-4-ylamino)pent-3-en-2-onate (L–). This is a fairly popular approach based on an acid-base type interaction. The proton transfer from “strong” acid LH to the coordinated anion (N(SiMe3)2)−, elimination of “weak” acid HN(SiMe3)2, and coordination of L− to the lanthanide cation occur as a result of the reaction. The reactions of Ln(N(SiMe3)2)3 with LH afforded compounds [Ln(L)3] isolated as crystalline phases of [Sm(L)3] · 0.5THF and [Gd(L)3] · 0.5C7H8 in 20 and 60% yields, respectively.
For unknown cause, the reproduction of the yield of Eu(N(SiMe3)2)3 synthesized using the standard procedure from EuCl3 and LiN(SiMe3)2 is unstable. Sometimes the yield does not exceed 10% [24]. To avoid this stage, another variant was used for the synthesis of the Eu(III) complex: the reaction of EuCl3 with LH in the presence of KN(SiMe3)2 as a strong base (Scheme 1). However, the yield of this reaction was very low: only several crystals of the target product [Eu(L)3] · 0.5THF were isolated upon concentrating the mother liquor.
The structures of all complexes were determined by X-ray diffraction analysis of single crystals. All compounds obtained are isotypical. The structures differ by different molecules of the solvate solvent: THF in the case of the Sm and Eu complexes and toluene for Gd. As compared to the [Zn(L)2] complex [18], the ligand in [Ln(L)3] · 0.5Solv is bound to the metal via another mode: the N atom of the heterocycle is involved in the coordination along with the N and O atoms of the ketoiminate moiety (Fig. 1a). Thus, L– is a tridentate ligand in these compounds. The coordination number of the central atom is 9, and the nearest environment of Ln can be described by a three-capped trigonal prism with three additional vertices above the lateral faces (Fig. 1b) or by a square one-capped antiprism with one additional vertex above the base (Fig. 1c).

(a) Structure of compounds [Ln(L)3 ] · 0.5Solv for Ln = Sm as an example; ellipsoids are presented with 50% probability; disordering, hydrogen atoms, and solvate molecules are omitted. (b, c) The nearest coordination environment of Ln.
The Ln–N and Ln–O bond lengths vary in wide ranges of 2.58–2.77 and 2.28–2.36 Å, respectively, which is characteristic of the lanthanide complexes (see further for an analysis of the bond lengths performed only for the fragments with the entire and for disordered atoms highest site occupancies). No relationships are observed between the Ln–N bond length and type of the nitrogen atom (thiadiazole moiety or imino group); i.e., the arrangement of the ligands relative to the central atoms is caused, to a considerable extent, by steric factors. Since Sm, Eu, and Gd have close ion radii, no significant differences are observed between the Ln–N and Ln–O bond lengths. Unlike the [ZnL2] complex with the planar structure of the metallocycle, the Ln atoms in [Ln(L)3] · 0.5Solv substantially deviate from the plane of the ketoiminate fragment (by 0.8 Å on the average). It is most likely that the distortion is related to different denticities of the ligand (bidentate in the Zn complex and tridentate in the Ln complexes). A specific feature of the studied compounds is the disordering of the Ln atom and one of three ligands over two positions (Fig. 2). Their refined site occupancy ranges from 92 : 8 to 84 : 16% in different crystals. The distance between the disordered Ln atoms does not exceed 0.62 Å. The corresponding coordinated heterocycles in the ligand also have close positions in pairs, but the O atom of one disordered fragment is localized near the N atom of the heterocycle of another fragment; i.e., these fragments are arranged according to the “head-to-tail” type (Fig. 2). In spite of this arrangement, the disordered fragments occupy almost identical volumes of the space, which results in failure of the molecular packing of the complexes. This disordering phenomenon was found for all samples of single crystals of the complexes.

Scheme of disordering of the central atom and ligand in compounds [Ln(L)3 ] · 0.5Solv for Ln = Sm as an example.
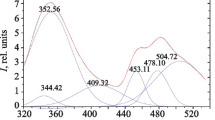
Thus, on the one hand, the thiadiazole moieties in the complexes are rather close to the central atoms, which is important for an efficient transfer of the excitation energy from the ligand to the lanthanide ions [29]. On the other hand, the color of the [Ln(L)3] · 0.5Solv complexes is bright red; i.e., the emission band of the Sm3+ and Eu3+ ions is too close in energy to the absorption band of the complexes and, as a consequence, the energy transfer from the ligand to these lanthanides followed by irradiation is poorly probable [29]. Indeed, no characteristic lanthanide-centered luminescence is observed in the emission spectrum of [Sm(L)3] · 0.5THF, but only the broad band assigned to the intraligand transitions is present (Fig. 3). The maximum of this band at 625 nm is shifted to the long-wavelength range compared to free LH (595 nm). It is important that the [Zn(L)2] complex with the bidentate coordination mode of the ligand performed by the atoms of the ketoiminate fragment only is yellow: the position of the long-wavelength absorption band of this compound is nearly identical with that of the initial LH [18]. It can be concluded that the bathochromic shift of the long-wavelength band of the [Ln(L)3] · 0.5Solv complexes is determined by the bond of the metal with the N atom of the heterocycle, which is absent from [Zn(L)2]. A similar phenomenon is observed in a series of the Zn, Cd, and Ag complexes with archetypal 4-amino-2,1,3-benzothiadiazole [18–21]. When this ligand is coordinated via the monodentate mode by the N atom of the amino group, the absorption and emission band is shifted to the short-wavelength range. When the ligand is coordinated via the monodentate mode by the N atom of the heterocycle or via the bridging mode, this band is shifted to the long-wavelength range. This assumption is consistent with both the experimental data and quantum chemical calculation results [20].

Normalized photoluminescence spectra of the solid samples of LH and [Sm(L)3] · 0.5THF.
To conclude, the new complexes with 4-(2,1,3-benzothiadiazol-4-ylamino)pent-3-en-2-onate [Ln(L)3] · 0.5Solv (Ln = Sm, Eu, Gd; Solv = THF, C7H8) were synthesized in this work. Their structures were determined by X-ray diffraction analysis. The disordering phenomenon due to close volumes of the ketoimine and benzothiadiazole moieties was discovered in the structures. The relationship between the coordination mode of the ligand and the long-wavelength band position in the electronic spectra of the complexes with L– was found.
REFERENCES
Neto, B.A.D., Lapis, A.A.M., da Silva Júnior, E.N., et al., Eur. J. Org. Chem., 2013, p. 228.
Gu, C., Liu, D., Wang, J., et al., J. Mater. Chem. A, 2018, vol. 6, p. 2371.
Crossley, D.L., Urbano, L., Neumann, R., et al., ACS Appl. Mater. Interfaces, 2017, vol. 9, p. 28243.
Chulanova, E.A., Pritchina, E.A., Malaspina, L.A., et al., Chem.-Eur. J., 2017, vol. 23, p. 852.
Lonchakov, A. and Rakitin, O., Gritsan, N., et al., Molecules, 2013, vol. 18, p. 9850.
Xu, Z., Kong, L., Wang, Y., et al., Org. Electron., 2018, vol. 54, p. 94.
Neto, B.A.D., Carvalho, P.H.P.R., and Correa, J.R., Acc. Chem. Res., 2015, vol. 48, p. 1560.
da Cruz, E.H.G., Carvalho, P.H.P.R., Correa, J.R., et al., New J. Chem., 2014, vol. 38, p. 2569.
Mota, A.A.R., Corrêa, J.R., Carvalho, P.H.P.R., et al., Org. Chem., 2016, vol. 81, p. 2958.
Aguiar, L.D.O., Regis, E., Tuzimoto, P., et al., Liq. Cryst., 2018, vol. 45, p. 49.
Langis-Barsetti, S., Maris, T., and Wuest, J.D., Org. Chem., 2017, vol. 82, p. 5034.
Cheng, Q., Han, X., Tong, Y., et al., Inorg. Chem., 2017, vol. 56, p. 1696.
Plebst, S., Bubrin, M., Schweinfurth, D., et al., Z. Naturforsch., A: Phys. Sci., 2017, vol. 72, p. 839.
Goswami, S., Winkel, R.W., and Schanze, K.S., Inorg. Chem., 2015, vol. 54, p. 10007.
Mancilha, F.S., Barloy, L., Rodembusch, F.S., et al., Dalton Trans., 2011, vol. 40, p. 10535.
Gallardo, H., Conte, G., Bortoluzzi, A.J., et al., Inorg. Chim. Acta, 2011, vol. 365, p. 152.
Gallardo, H., Conte, G., Tuzimoto, P., et al., Inorg. Chem. Commun., 2008, vol. 11, p. 1292.
Sukhikh, T.S., Bashirov, D.A., Ogienko, D.S., et al., RSC Adv., 2016, vol. 6, p. 43901.
Sukhikh, T.S., Ogienko, D.S., Bashirov, D.A., et al., J. Coord. Chem., 2016, vol. 69, p. 3284.
Sukhikh, T.S., Komarov, V.Y., Konchenko, S.N., et al., Polyhedron, 2018, vol. 139, p. 33.
Sukhikh, T.S., Bashirov, D.A., Shuvaev, S., et al., Polyhedron, 2018, vol. 141, p. 77.
Sukhikh, T.S., Bashirov, D.A., Kolybalov, D.S., et al., Polyhedron, 2017, vol. 124, p. 139.
Sukhikh, T.S., Bashirov, D.A., Kuratieva, N.V., et al., Dalton Trans., 2015, vol. 44, p. 5727.
Herrmann, W.A., Synthetic Methods of Organometallic and Inorganic Chemistry: Lanthanides and Actinides, Thieme Publishing Group, 1997, vol. 6.
APEX2 (version 2.0). SAINT (version 8.18c), and SAD-ABS (version 2.11), Madison: Bruker Advanced X‑ray Solutions, 2000−2012.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, p. 112.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Armelao, L., Quici, S., Barigelletti, F., et al., Coord. Chem. Rev., 2010, vol. 254, p. 487.
ACKNOWLEDGMENTS
The authors are grateful to A.P. Zubareva and O.S. Koshcheeva for performing elemental analysis and to A.A. Ryadun for recording luminescence spectra.
This work was carried out in terms of the state task and was supported by the Russian Foundation for Basic Research, project no. 16-03-00637.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Sukhikh, T.S., Ogienko, D.S., Bashirov, D.A. et al. Samarium, Europium, and Gadolinium Complexes with 4-(2,1,3-Benzothiadiazol-4-ylamino)pent-3-en-2-onate. Russ J Coord Chem 45, 30–35 (2019). https://doi.org/10.1134/S1070328419010111
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328419010111