
Abstract
Objective: Structural analysis of the fatty acids and their ethyl esters from the extract of sponge Penares sp. (South China Sea). Methods: Separation by high-performance liquid chromatography. Analysis by gas chromatography-mass spectrometry using pyrrolidine, 4,4-dimethyloxazoline, dimethyl disulfide, and hydrogenated derivatives. Analysis by 1Н and 13С NMR spectroscopy. Results and Discussion: 71 acids with a chain length from C12 to C28 were found, including 12 new compounds, i.e., (5Z,9Z)-9-chloro-24-methy-5,9-pentacosadienoic, (5Z,9Z)-9-chloro-25-methyl-5,9-hexacosadienoic, (5Z,9Z)-9-chloro-24-methyl-5,9-hexacosadienoic, (5Z,9Z)-9-chloro-25-methyl-5,9-heptacosadienoic, 6-chloro-20-methyl-4-heneicosenoic, 6-chloro-19-methyl4-heneicosenoic, 6-chloro-20-methyl-4-docosenoic, cis-17,18-methylene-tetracosanoic, 16,21-dimethyldocosanoic, 18,23-dimethyltetracosanoic, 16,18,22-trimethyltricosanoic, and 18,20,24-trimethylpentacosanoic acids. Conclusions: The characteristic features of the fatty acid mixture from Penares sp. were a high level of constituents with monomethylated chains (over 50%) and the nearly total substitution of common demospongic acids for their previously unknown chloro derivatives, (5Z,9Z)-9-chloro-5,9-dienoic acids, due to, presumably, the activity of sponge-associated microorganisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Sponges are ancient and the most primitive multicellular animals, which are one of the richest natural sources of various unique metabolites including unusual fatty acids (FAs). Many experimental and some review articles are devoted to sponge FAs (e.g., reviews [1–5]). Despite the predominance of chloride ions in seawater, the previously known halogenated sponge FAs are represented by 6-bromo derivatives of 5-cis,9-cisdienoic (or demospongic [6]) acids and acetylenic polyunsaturated brominated [7] and iodinated [8] acids. Chlorinated FAs were found in some more evolutionarily developed marine invertebrates, fish, and algae [7].
When separating the ethanol extract of sponge Penares sp. (South China Sea), we obtained not only fractions of triterpenoids [9] and brominated indole alkaloids [10], but also a fraction of ethyl esters of new demospongic acids that contained a vinylic chlorine atom. This unusual structural feature prompted us to investigate the FAs of this sponge in more detail, which led to the finding of other previously unknown acids.
Penares sponges are producers of many lipids and lipid-like compounds with various biological activities. For example, penaresidins A and B from Penares sp. activated actin-dependent ATPase [11], and penazetidine A from Penares sollasi inhibited protein kinase C [12]. Penasulfate A from Penares sp. and schulzeines A−C from Penares schulzei inhibited α-glucosidase [13, 14], and ancorinosides B–D from Penares sollasi inhibited membrane matrix metalloproteinase (MT1-MMP) [15]. Penaramides from Penares aff. incrustans inhibited the binding of ω-conotoxin GVIA to the N-type calcium channels [16], and penasins A–E from Penares sp. had cytotoxic activity against the HeLa and P388 tumor cells [17]. Despite the interest in these substances, a structural analysis of their potential precursors, i.e., individual FAs from the sponges of the genus Penares, was not previously performed. The only screening studies of FAs of two Penares tylotaster samples were published, and the relative FA contents of these samples were compared by chain lengths and the presence/absence of branching, double bonds, etc., without revealing the position of substituents and double bonds [18, 19]. The goal of our work was a more thorough structural analysis of FAs from a sponge, belonging to the genus Penares, and a comparison of the characteristic structural features of some detected FAs and known secondary metabolites of Penares.
RESULTS AND DISCUSSION
Fractions of FAs and their ethyl esters were isolated from the ethanol extract of Penares sp. by column chromatography on Sephadex and silica gel and separated by HPLC on normal-phase or reversed-phase columns or both. The resulting fractions were analyzed by GC-MS (electron impact ionization) using ester, pyrrolidine, 4,4-dimethyloxazoline, dimethyl disulfide, and hydrogenated FA derivatives. In some cases, 1H and 13C NMR spectroscopies were used to analyze FA structures. We found 71 acids with a chain length from C12 to C28 (Table 1) including 12 new compounds, i.e., (5Z,9Z)-9-chloro-24-methyl-5,9-pentacosadienoic (I), (5Z,9Z)-9-chloro-25-methyl-5,9-hexacosadienoic (II), (5Z,9Z)-9-chloro-24-methyl-5,9-hexacosadienoic (III), (5Z,9Z)-9-chloro-25-methyl-5,9-heptacosadienoic (IV), 6-chloro-20-methyl-4-heneicosenoic (V), 6-chloro-19-methyl-4-heneicosenoic (VI), 6-chloro-20-methyl-4-docosenoic (VII), cis-17,18-methylene-tetracosanoic (VIII), 16,21-dimethyldocosanoic (IX), 18,23-dimethyltetracosanoic (X), 16,18,22-trimethyltricosanoic (XI), and 18,20,24-trimethylpentacosanoic (XII) acids (Fig. 1). Previously unknown compounds (I‒XII) were observed in trace amounts (<0.1%) in the total FA mixture.

New fatty acids (FAs) from sponge Penares sp.
Structural Analysis of New FAs
Like the mass spectra of higher aliphatic monohalides [20], the mass spectra of the ethyl esters (Ia–IVa) of chlorinated demospongic acids contained either no or very low-intensity peak of molecular ion [M]+ because of a slight loss of Cl and HCl. In contrast, we observed relatively intense peaks of [M ‒ Cl]+/[M ‒ HCl]+/[M ‒ Cl ‒ EtOH]+ ions (m/z 419/418/373, 433/432/387, and 447/446/401 for the ethyl esters of C26, C27, and C28 acids, respectively). Noticeable amounts of [M – 88 – Cl]+ ions, where the fragment with m/z 88 was formed due to the McLafferty rearrangement [21], were also detected. The intense peaks at m/z 55, 67, 81 (100%), 95, and 109, and some other peaks, typical for the fragmentation of the ethyl esters of dienoic FAs [22], were observed in the region of low ion masses. In addition to the peak at m/z 155, characteristic of the mass spectra of Δ5,9-acid ethyl esters [23], a slightly more intense peak at m/z 154 was observed in the mass spectra of compounds (Ia–IVa). The double signal at m/z 154/155 was accompanied by less intense signals of two long-chain isotopic ions with 35Cl and 37Cl. These ions containing the methyl end of the molecule were also formed by the cleavage of the bis-allylic bond CH2-7–CH2-8 (Fig. 2a). Thus, in the mass spectra of the ethyl esters of C26, C27, and C28 acids, two isotopic peaks at m/z 298/300, 312/314, and 326/328, respectively, in a characteristic ratio of ~3 : 1 were observed. The summation of the masses of each of the two isotopic ions with a sum value of 155 + 1 gave the corresponding molecular masses, e.g., 454/456 [M]+ for (Ia). In addition, the signals of the secondary radical ions with m/z 403 ([M ‒ HCl – CH3]+ for (Ia) or [M ‒ HCl – CH2CH3]+ for (IIIa)) and m/z 417 ([M ‒ HCl – CH3]+ for (IIa) or [M ‒ HCl – CH2CH3]+ for (IVa)) were more intense than neighboring signals of homologous ions, which implied the presence of iso- or anteiso-branching in the corresponding structures (e.g., Fig. 2a).

Mass spectrometric fragmentation of isomeric ethyl esters (IIa) and (IIIa) (the peak of the molecular ion at m/z 468 [M]+ with 35Cl was low-intensity and the minor peak [M]+ with 37Cl was not recorded) (a); mass spectrometric fragmentation of the bis(methylthio) derivative of ethyl ester (Ia) (513 [M – Cl]+) (b); mass spectrometric fragmentation of pyrrolidide (Ib) (479/481 [M]+; to simplify the scheme, less abundant ions corresponding to the elimination of HCl from the isotopic fragments at m/z 310/312–422/424) are not shown (c); key НМВС correlations for compounds (IIa) and (IIIa) (d).
Compounds (Ia–IVa) were converted to dimethyl disulfide derivatives. In this case, dimethyl disulfide was not attached to the chloro-substituted double bonds of (Ia–IVa). The mass spectra of the resulting adducts contained the significant peak of [M – Cl]+ ion ([M]+ ion was not detected) and the intense peaks at m/z 129 (100%) and 175, thus indicating the presence of double bond at position 5 of the initial compounds. The latter signals were accompanied by much less intense signals of isotopic ions, which were also formed by the cleavage of the C5–/–C6 bond, e.g., the ions of m/z 373/375 produced by fragmentation of the bis(methylthio) derivative of ethyl ester (Ia) (Fig. 2b). The summation of the masses of the isotopic m/z 373/375 ions with the mass of m/z 175 ion gave the values of 548/550 [M]+ for the dimethyl disulfide derivative of (Ia).
The mass spectra of pyrrolidides (Ib–IVb) and 4,4-dimethyloxazoline derivative of acid (I) contained the noticeable peaks of isotopic molecular ions (m/z 479/481, 493/495, and 507/509 with 35C1/37C1 for the derivatives of C26, C27, and C28 acids, respectively) along with slightly more intense peaks of [M ‒ Cl]+ ions. The intense peak of m/z 180 ion, which was formed by the cleavage at the center of the bis-methylene-separated system of two double bonds (Fig. 2c), unambiguously indicated the presence of Δ5,9 fragment in all analyzed N-containing derivatives [23, 24]. The positions of methyl branches in acids (I–IV) were revealed by gaps between certain peaks in the mass spectra of their pyrrolidides according to the previously formulated rule [25]. For example, the mass spectra of pyrrolidides (Ib) and (IIIb) with methyl group at position 24 had the gap of 28 amu between the peaks at m/z 464/466 ([M ‒ CH3]+ for (Ib) and [M ‒ CH2CH3]+ for (IIIb)) and 436/438 ([M ‒ CH(CH3)2]+ for (Ib), and [M ‒ CH(CH3)CH2CH3]+ for (IIIb)). Fragmentation of pyrrolidide (IIb) that contained methyl group at CH-25 resulted in the gap of 28 amu between the peaks at m/z 478/480 ([M ‒ CH3]+) and 450/452 ([M ‒ CH(CH3)2]+). The signal of the secondary radical ion of m/z 478/480 [M – CH2CH3]+ in the mass spectrum of pyrrolidide (IVa) was also much more intense than the neighboring signals of homologous ions, which indicated the presence of methyl group at CH-25. The mass spectra of pyrrolidides demonstrated the break of sequential fragmentation after CH2-8 and its continuation after CH-10 as shown for (Ib) (Fig. 2c). The same pattern was observed in the mass spectrum of 4,4-dimethyloxazoline derivative of acid (I) (478/479/480/481, [M – 1]+/[M]+), which was isomer of pyrrolidide (Ib). Thus, the mass spectrometric data indicated that chlorine atom in compounds (I–IV) could be located at either C9 or C10.
Hydrogenation of some derivatives of acids (I–IV) over the Adams catalyst was accompanied by dehalogenation, thus leading to the transformation of these substances into known saturated compounds. For example, pyrrolidide (Ib) was dehalogenated to the corresponding derivative of 24-methylpentacosanoic acid, the mass spectrum of which contained a diagnostic gap (28 amu) between the peaks at m/z 406 and 434 [26]. Ethyl esters (IIa–IVa) were transformed during hydrogenation into the ethyl esters of 25-methylhexacosanoic, 24-methylhexacosanoic, and 25-methylheptacosanoic acids (equivalent chain lengths (ECL) were 26.63, 26.72, and 27.72, respectively, according to the GC-MS analysis). Elimination of Cl during hydrogenation could indicate its vinylic (as in compounds (I–IV)) or allylic (as in compounds (V–VII)) location because dehalogenation of saturated chlorinated fatty acids did not occur under the used hydrogenation conditions.
Using HPLC on normal-phase and reversed-phase columns, we managed to obtain a mixture of structurally similar isomers (IIa) and (IIIa) (39 and 31% in the mixture, respectively), which were not separated by preparative chromatography. The presence of the 5,9-diene fragment and the position of the chlorine atom at C9 in these compounds were confirmed by 1H,1H-COSY, HMBC, and HSQC experiments. The 1H,1H-COSY spectrum of compounds (IIa) and (IIIa) showed the presence of linear spin proton systems from CH2-2 (δH 2.30, t; J 7.5) to CH2-8 (δH 2.325, t; J 7.3) and from CH-10 (δH 5.44, t; J 7.0) to CH2-pool (δH 1.20–1.35, m). The HMBC spectrum of (IIa) and (IIIa) contained the corresponding correlations (Fig. 2d). The value of the spin-spin coupling constant J5,6 10.9 Hz, which was found by selective homonuclear decoupling of allylic protons [27], indicated Z-configuration of the double bond at C5. Z-Configuration of the double bond at C9 was revealed by the comparison of the 1H and 13C NMR spectra of compounds (IIa) and (IIIa) with the corresponding literature spectra of some known vinylically chlorinated Z/E isomers [28], i.e., by the presence of characteristic resonances of CH-10 (δH 5.44, t; J 7.0), CH2-11 (δH 2.145, m), and CH2-8 (δC 39.4). In addition, the NMR spectra showed the signals of two equivalent terminal methyl (δH 0.86, d; J 6.7; δC 22.6), one methine (δH 1.515, m; δC 28.0), and one methylene (δH 1.15, m; δC 39.0) groups, the protons of which formed the spin system of the isostructure of component (IIa) according to 1H,1H-COSY and HMBC correlations. The correlations in the 1H,1HCOSY and HMBC spectra made it possible to confirm the anteiso-structure of component (IIIa), which showed the characteristic signals of two methyl groups (δH 0.84, d; J 6.2; δC 19.2 and δH 0.85, t; J 7.1; δC 11.3) [29, 30].
The values of chemical shifts and the multiplicities of olefin proton signals in the 1H NMR spectra of HPLC fractions enriched with acids (I) and (IV) were analogous to the corresponding characteristics of the protons of double bonds in the ethyl esters of acids (II) and (III). This fact and data from mass spectrometric fragmentation and chemical transformations of (I–IV) indicated that these compounds contained identical 5Z,9Z-diene bond system with chlorine atom at C9. This conclusion was confirmed by the ECL values (27.00 and 28.00 for isohomologues (I) and (II), respectively, and 28.11 and 29.12 for anteiso-homologues (III) and (IV), respectively).
Allylic halides are known to lose halogen more easily than vinylic halides. This pattern was manifested under the conditions of mass spectrometric fragmentation of allylically chlorinated pyrrolidides (Vb–VIIb), for which the elimination of chlorine atom occurred easier than for vinylically chlorinated pyrrolidides (Ib–IVb). For example, the main signal in the mass spectra of pyrrolidides (Vb–VIIb) was the peak of [M – Cl]+ ion (m/z 390, 390, and 404, respectively), but the intensity of this peak in the mass spectra of pyrrolidides (Ib–IVb) was only ~3.5%. The molecular ion region in the mass spectra of compounds (Vb‒VIIb) contained clusters of low-intensity peaks of isotopic ions [M – 1]+/[M]+ (m/z 424/425/426/427, 424/425/426/427, and 438/439/440/441, respectively). Apart from the peak at m/z 126, the region of low ion masses contained much less intense peaks at m/z 138/139 and 152, which was a sign of the presence of a double bond at C4 (Fig. 3a). Although a clear signal of allylic cleavage at m/z 166 was characteristic of the mass spectra of the pyrrolidides of known Δ4 acids [26], the mass spectra of the pyrrolidides of Δ4 acids (Vb–VIIb) were characterized by an increased intensity signal of a similar mass ion of m/z 165. Obviously, this ion was formed because of the abstraction of chlorine atom from the initial products of allylic cleavage, i.e., isotopic ions with m/z 200 and 202, which were detected in the mass spectrum as less intense peaks in the ratio of 3 : 1 despite the easy loss of Cl during fragmentation. This kind of characteristic cleavage after substituted allylic carbon, which led to the appearance of significant diagnostic signals, was previously observed during the mass spectrometric fragmentation of the methyl esters of some FAs with allylic hydroxy or S-methyl groups [31]. The gap of 28 amu between the peaks at m/z 374 (corresponds to the [M ‒ CH3 – HCl]+ ion for (Vb) and the [M ‒ CH2CH3 – HCl]+ ion for (VIIb)) and 346 ([M ‒ CH(CH3)2 – HCl]+ for (Vb) and [M ‒ CH(CH3)CH2CH3 – HCl]+ for (VIIb)) indicated the presence of the methyl group at C20 in acids (V) and (VII). This conclusion was confirmed by the increased intensity of the peaks of the secondary ion radicals at m/z 410/412, which represented a fragment [M ‒ CH3]+ for iso-methyl-branched compound (Vb) and a fragment [M ‒ CH2CH3]+ for anteiso-methyl-branched compound (VIIb). The gap of 28 amu between the peaks at m/z 360 ([M ‒ CH2CH3 – HCl]+) and 332 ([M ‒ CH(CH3)CH2CH3 – HCl]+) and the peak of increased intensity of the secondary ion radical at m/z 396/398 [M ‒ CH2CH3]+ in the mass spectrum of anteiso-methyl-branched pyrrolidide (VIb) were signs of the presence of methyl group at C19. Expectedly, hydrogenation of compounds (Vb–VIIb) over the Adams catalyst proceeded with dehalogenation, thus leading to the formation of the pyrrolidides of known 20-methylheneicosanoic, 19-methylheneicosanoic, and 20-methyldocosanoic acids, which we also found in Penares sp. Unfortunately, the low content of allylically chlorinated acids (V–VII) made it impossible to register the 1H NMR spectrum, which would be quite informative to establish the configuration of the double bond of these compounds.

Mass spectrometric fragmentation of pyrrolidide (Vb) (424/425/426/427 [M – 1]+/[M]+) (a); mass spectrometric fragmentation of ethyl esters (XIa) and (XIIa) (b).
The peaks of ions with m/z 408 [M]+, 362 [M – EtOH]+, etc., were detected in the mass spectrum of the ethyl ester of cyclopropane-containing acid (VIII) similar to those for monoenoic C25 acid. However, the inability of the ethyl ester of compound (VIII) to attach dimethyl disulfide and its ECL value (24.94) implied the presence of the cyclopropane ring at the (n –7) and (n –8) carbon atoms (n is the sequence number of the terminal carbon atom of the linear aliphatic chain) as in the ethyl esters of homologous 9,10-methylene-16:0 (ECL 16.94) and 11,12-methylene-18:0 (ECL 18.93) acids from Penares sp. (Table 1). The mass spectrometric features of the pyrrolidides of higher cyclopropane-containing homologs (C19 and more) of such acids include an intense peak with an odd m/z value corresponding to the fragment ion, which is formed by β-cleavage to the cyclopropane ring, and an interval of 12 amu between the fragments showing the position of the cycle [32, 33]. Accordingly, the mass spectrum of the pyrrolidide of acid (VIII) (m/z 433 [M]+) contained an intense peak at m/z 363 (ion formed by β-cleavage) and a characteristic interval of 12 amu between the peaks at m/z 322 and 334 corresponding to C17 and C18 FA fragments, which confirmed the presence of the cyclopropane ring in the 17,18 position. In the 1H,1HCOSY spectrum, protons with δH −0.33 (ddd, J 4.3, 5.3, 5.3, 1H, H-25a), 0.56 (ddd, J 4.3, 8.4, 8.4, 1H, H-25b), and 0.65 (m, 2H, H-17, H-18) of acid (VIII) were coupled to each other within a three-membered cycle; in this case, the values of chemical shifts and spin-spin coupling constants for the signals of these protons corresponded to the cis-orientation of the ring [33, 34].
It should be noted that the mass spectra of the studied 4,4-dimethyloxazoline derivatives of cyclopropanecontaining FAs are considered almost indistinguishable from the mass spectra of 4,4-dimethyloxazoline derivatives of unbranched monoenoic FAs of the same molecular weight because of the probable rearrangement of cyclopropanes into monoenes during fragmentation [35, 36]. According to this pattern, the mass spectrum of 4,4-dimethyloxazoline, formed from acid (VIII), should be similar to the mass spectrum of 4,4-dimethyloxazoline, formed from acid 25:1Δ17. Nevertheless, the fragmentation of obtained 4,4-dimethyloxazoline derivative of acid (VIII) (m/z 433 [M]+) resembled the fragmentation of the corresponding derivative of acid 25:1Δ18 because it led to the appearance of an interval of 12 amu between the peaks at m/z 322 (C17 fragment) and 334 (C18 fragment). In this case, these peaks were even slightly more intense than the previous peak at m/z 308, which did not correspond to the relative intensity of the signals, characteristic of fragmentation near the isolated double bond. Thus, in the recorded mass spectrum of the 4,4-dimethyloxazoline derivative of acid (VIII), the characteristic signals with an interval of 12 amu were shifted and changed their intensities contrary to the pattern previously observed in the mass spectra of 4,4-dimethyloxazoline derivatives of shorter cyclopropane-containing FAs. Similar changes in the diagnostic signals as a result of increasing aliphatic chain length were previously observed in the mass spectra of pyrrolidides of some homologous cyclopropanecontaining FAs, which, like compound (VIII), were formal derivatives of monoenes of the (n –7) family [33].
The ethyl esters of two saturated homologous isomethyl-branched acids (IX) (ECL 22.97) and (X) (ECL 24.98) with additional methyl group at the (n –7) position generated characteristic ions formed by the cleavage of the bonds in the α positions relative to the (n –7) carbon as previously described for the mass spectrometric fragmentation of the FA esters with methyl group in the middle of chain [37]. For example, unlike the ethyl ester of standard unbranched 24:0 acid (m/z 396 [M]+), the α-cleavage of the ethyl ester of compound (IX) (m/z 396 [M]+) produced the ions with m/z 269 and 297 in an approximately equal ratio and the fragments with m/z 251 and 233 because of the abstraction of the EtOH molecule and the EtOH and H2O molecules, respectively, from the ion with m/z 297. The ethyl ester of compound (X) (m/z 424 [M]+) generated large amounts of the ions at m/z 325 and 297, compared to the corresponding derivative of unbranched 26:0 acid (m/z 424 [M]+), and the ions at m/z 279 (the abstraction of the EtOH molecule from the ion with m/z 325) and 261 (the abstraction of the EtOH and H2O molecules from the ion with m/z 325). In this case, the signals of the fragments indicating the presence of the terminal iso-structures in the ethyl esters of acids (IX) and (X) were low-intense. The positions of all methyl branches were unambiguously determined based on the presence of the diagnostic gaps of 28 amu between the peaks at m/z 294 and 322 (loss of CH-16 with its methyl group) and the peaks at m/z 378 and 406 (loss of CH-21 with its methyl group) in the mass spectrum of the pyrrolidide of 16,21-dimethyl-branched acid (IX) (m/z 421 [M]+), as well as the similar gaps between the peaks at m/z 322 and 350 (loss of CH-18 with CH3) and m/z 406 and 434 (loss of CH-23 with CH3) in the mass spectrum of the pyrrolidide of 18,23-dimethyl-branched acid (X) (m/z 449 [M]+).
iso-Methyl-branched homologous acids (XI) (ELC 24.53) and (XII) (ECL 26.53) had additional methyl groups at the (n –6) and (n –8) positions. Figure 3b shows some fragment ions that gave increased intensity signals in the mass spectra of ethyl esters (XIa) (m/z 424 [M]+) and (XIa) (m/z 452 [M]+) compared with the mass spectra of unbranched standards of similar molecular weights. In addition, the characteristic ions that were absent in the mass spectra of the standards, are shown in Fig. 3b. It can be seen that the fragmentation near C16 in compound (XIa) and C18 in compound (XIIa) was similar to the fragmentation of the ethyl esters of acids (IX) and (X), respectively. The detection of the gaps of 28 amu between the peaks at m/z 294 and 322, 336 and 364, and 406 and 434 in the mass spectrum of the pyrrolidide of acid (XI) (m/z 449 [M]+) indicated the presence of the methyl groups at C16, C18, and C22. The gaps between the peaks at m/z 322 and 350, 364 and 392, and 434 and 462 in the mass spectrum of the pyrrolidide of acid (XII) (m/z 477 [M]+) corresponded to the positions of the methyl branches at C18, C20, and C24.
Features of Fatty Acids from Penares sp.
Most fatty acids in living tissues exist in a bound form (as components of other lipids), whereas free or non-esterified FAs with harmful properties are minor compounds. However, these compounds are easily generated from more complex lipids during storage (even at –20°C), thawing, or extraction of biological objects due to the functioning of lipases [38]. Therefore, we consider FAs isolated from the Penares sp. extract as products of the activity of these enzymes. As for the ethyl esters of FAs, these compounds are, in our experience, common components of ethanol sponge extracts.
The FA mixture from Penares sp. (Table 1) contains higher amounts of branched components (59.1%) than the FA mixture of previously studied Penares tylotaster (38.1%) [19]. The branched C15–C28 acids from Penares sp. have mono-, di-, and tri-methylated chains. Among the dominant monomethylated FAs, there are iso/anteiso components (12.4/9.6%) and components with mid-chain methyl group (37.1%) located at positions (n –5), (n –8), (n –6), and (n –7) of the aliphatic chain (in order of relative abundance). Most monomethylated acids have an odd number of carbon atoms, i.e., C19 (17.1%; isomeric 10-Me-18:0 and 11-Me-18:0 prevail), C17 (12.4%), C25 (10.4%), and C15 (9.4%). Di- and trimethylated FAs are trace components. The source of methyl-branched and cyclopropane-containing FAs of sponges are considered to be bacteria that serve as their food or inhabit these invertebrates [3, 39–41]. Some sponges, like “microbial fermenters,” can contain so many bacteria per gram of weight that they have even been called “bacteriosponges” [42]. The FAs of bacteria and other associated microbes can be present in unchanged form in fatty acid fractions of sponges or be exogenous precursors of longer FAs of these invertebrates (review by Bergé et al. [3] and references therein). Consequently, bacterial 9,10-methylene-16:0 or 11,12-methylene-18:0 acids, which were found in minor amounts in Penares sp., could act as an elongation substrate during the formation of compound (VIII). The precursors of dimethylated (IX and X) and trimethylated (XI and XII) compounds could also be shorter homologs of bacterial origin, such as 8-Me-iso-15:0 and 10,12-di-Me-iso-18:0 acids, respectively, which were previously found in bottom sediments [43]. It should be noted that methyl branches are characteristic of the aliphatic chains of many biologically active lipids and lipid-like compounds isolated from sponges of the Penares genus including penaresidins A and B (iso- and anteiso-branching) [11], penazetidine A (methyl group at the (n –7) position) [12], penasulfate A (methyl group in the (n –7) position) [13], schulzeine A (methyl group at the (n –9) position) [14], penaramides (iso- and anteiso-branching or methyl group at the (n –7) position) [16], penasins C–E (methyl groups at the (n –8), (n –9), or (n –10) positions) [17], and ancorinoside C (methyl group in the middle of glycosylated chain) [15]. These structural features may not only imply the involvement of bacteria in the biosynthesis of these compounds but also suggest a significant amount of these microorganisms in sponges of the Penares genus.
Saturated linear FAs of Penares sp. (Table 1) contains С12, С14–С20, and С22–С26 components (36.0% with the predominance of 16:0 (8.9%) and 18:0 (8.4%) acids). Linear monoenes (14.2%) are 18:1Δ9 (13.4%) and 16:1Δ9 (0.8%) acids. A demospongic acid with unbranched skeleton, 26:2Δ5Z,9Z, is present in trace amounts. These saturated, monoenoic and dienoic FAs are typical for many sponges [2, 5]. The remaining unsaturated FAs of Penares sp. include previously unknown chlorinated acids with a methyl branch at the end of their aliphatic chains, i.e., monoenes: 6-Cl-iso-22:1Δ4 (V), 6-Cl-anteiso22:1Δ4 (VI), and 6-Cl-anteiso-23Δ4 (VII) and dienes: 9-Cl-iso-26:2Δ5Z,9Z (I), 9-Cl-iso-27:2Δ5Z,9Z (II), 9-Clanteiso-27:2Δ5Z,9Z (III), and 9-Cl-anteiso-28:2Δ5Z,9Z (IV). Thus, demospongic acids, typical for sponges, were almost completely replaced by their chlorinated derivatives.
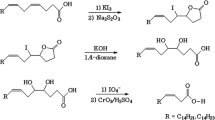
Chlorination of natural fatty acids occurs under the action of halogenating enzymes, e.g., chloroperoxidases and chlorinating halogenases [44]. These enzymes could convert 5Z,9Z-dienoic acids of Penares sp. into corresponding 9-chloro-acids (I–IV), the first representatives of chlorinated demospongic acids. Cytotoxic penasin A (XIII) that contains bis-methylene-separated cis-double bonds and its chlorinated derivative, penasin B (XIV), from a Japanese sample of a sponge of the Penares genus [17] can be considered as a substrate and a product of analogous chlorination (Fig. 4). The simultaneous presence of triterpenoid (XV) and its chlorohydrin derivative (XVI) in the sample of Penares sp. (Fig. 4) (the structures of these compounds were established earlier [9]) can also be considered a sign of the functioning of halogenating enzyme, which promotes the production of hypochlorous acid HClO. It is likely that the enzyme-catalyzed addition of halogen to C6 in Δ5-acids common to many marine sponges [2, 5] could cause a double bond shift by analogy with lipoxygenase hydroperoxidation and lead to the formation of allyllically chlorinated Δ4-acids (V–VII).

Hypothetical pathways for the formation of chlorinated secondary metabolites (XIV) and (XVI) isolated from sponges of the Penares genus.
Halogenating enzymes were not detected in sponge cells. However, the genes of halogenases (which belong to some bacteria according to some signs) were found in the microbial metagenomes from sponges with a high content of microorganisms [45]. The proportion of the components of bacterial origin in the studied mixture of FAs from Penares sp. made it possible to conclude that the bacterial content in this sponge was high, which could ensure the halogenation of FAs and other metabolites. It is also possible that not only sponge-associated bacteria (including cyanobacteria or blue-green algae) but also fungi could be responsible for this halogenation [46]. In the case of Penares sp., microbial chlorinating enzymes obviously preferred the double bond-containing aliphatic chains as a substrate, thus leading to the formation of acids (I–VII). Apart this, brominating enzymes were also active in Penares sp. studied here because, in addition to triterpenoids, indole alkaloids containing bromine in aromatic rings were previously isolated from this sample [9, 10]. Obviously, despite the functioning of bromination mechanism, microbes from Penares sp. used chlorination rather than bromination of demospongic acids in contrast to microorganisms associated with other sponges.
EXPERIMENTAL
Equipment. The 1H NMR, 1H,1H-COSY, HSQC, and HMBC (CDCl3) spectra were recorded on Avance III HD 500 (Bruker, Germany, 500.13 MHz) and Avance III 700 (Bruker BioSpin, Germany,700.13 MHz) spectrometers using tetramethylsilane as the internal standard.
The GC-MS analysis was performed on an HP6890 GC System chromatograph-mass spectrometer (Hewlett-Packard, USA) using an HP-5MS capillary column (30.0 m × 0.25 mm; J&W Scientific, USA), helium as a carrier gas, and an ionizing potential of 70 eV. In most cases, we used the following program: 100°C (1 min)— 10°C/min—280°C (30 min) at the injector temperature of 270°C. For the low-volatile pyrrolidides of longchain FAs, we used the following program: 200°C (1 min)—30°C/min—280°C (45 min) at the injector temperature of 300°C. HPLC was performed on liquid chromatographs, i.e., 1) Du Pont Series 8800 Instrument (DuPont, USA) equipped with RIDK-102 refractometer (Laboratorni Pristroje, Czechoslovakia) and ZORBAX Eclipse XDB-C8 column (4 × 150 mm; Agilent Technologies, USA) in 85% ethanol; 2) Agilent 1100 Series (Agilent Technologies, USA) equipped with RID-G1362A differential refractometer on ULTRASPHERATM Si column (10 × 250 mm; Beckman Instruments, USA) in a petroleum ether–ethyl acetate mixture (100 : 1) and YMC-Pack ODS-A column (10 × 250 mm; YMC Co., Japan) in ethanol.
Column chromatography was performed on Sephadex LH-20 (Sigma Chemical Co., USA) and silica gel (50/100 or 50/160 µM; Sorbpolymer, Russia). The qualitative analysis was performed using thin-layer chromatography on Sorbfil plates (Sorbpolymer, Russia) with a layer of STX–1A silica gel fixed to the foil. The spots of the substances were stained by spraying with an EtOH-H2SO4 mixture (1 : 1).
Biological material. A sample of a sponge of the Penares genus was collected by dredging from a depth of 95 m in the South China Sea (16°07'N, 114°47'E) in January 2005 during the 30th voyage of the Academic Oparin research vessel to Vietnam. The sponge was identified by V.B. Krasokhin (G.B. Elyakov Pacific Institute of Bioorganic Chemistry, FEB RAS, Vladivostok, Russia). The sample (PIBOC O30-271) is stored in the collection of Elyakov Pacific Institute of Bioorganic Chemistry, FEB RAS.
Sponge extraction and separation of FA fractions and FA ethyl esters. The freshly harvested Penares sponge (400 g) was frozen and stored at –20°C. The sponge was crushed and extracted with ethanol at room temperature. The ethanol extract was concentrated in a vacuum to an aqueous residue, which was extracted with hexane (3 × 250 mL). The hexane extract (2.92 g) that contained nonpolar and low-polar sponge compounds was divided into fractions on a Sephadex LH-20 column using a CHCl3–EtOH mixture (1 : 1) as an eluent. The resulting fraction (346.4 mg) contained similar-sized molecules of FAs and their ethyl esters. These compounds were separated according their different polarity on a silica gel column. The substances (26.9 mg), which were eluted in hexane-ethyl acetate (70 : 1), were separated by normalphase HPLC (petroleum ether-ethyl acetate, 100 : 1) and reversed-phase HPLC (ethanol). We obtained 1.5 mg of a mixture of ethyl esters (IIa) and (IIIa). The substances (147.9 mg), which were eluted in hexane-ethyl acetate (5 : 1 → 2 : 1) from silica gel, were separated by reversedphase HPLC (85% ethanol) to obtain FA fractions.
Ethyl esters (IIа) and (IIIа) (39 and 31% in the HPLC fraction, respectively); colorless oily substance; 1Н NMR (CDCl3, 700 MHz; δ, ppm (J, Hz)): 5.44 (t, J 7.0, H-10), 5.37 (m, Н-5), 5.36 (m, H-6), 4.13 (q, J 7.1, −OCOСН2CH3), 2.325 (t, J 7.3, CH2-8), 2.30 (t, J 7.5, CH2-2), 2.27 (m, CH2-7), 2.145 (m, CH2-11), 2.09 (m, CH2-4), 1.69 (m, CH2-3), 1.515 (m, Н-25, (IIа)), 1.35–1.20 (m, CH2-pool), 1.32 (m, H-25b, (IIIа)), 1.30 (m, H-24, (IIIа)), 1.26 (m, Н-23b, (IIIа)), 1.255 (t, J 7.1, −OCOСН2CH3), 1.15 (m, СН2-24, (IIа)), 1.12 (m, Н-25а, (IIIа)), 1.08 (m, Н-23а, (IIIа)), 0.86 (d, J 6.7, CH3-27, 26, (IIа)), 0.85 (t, J 7.1, CH3-26, (IIIа)), and 0.84 (d, J 6.2, CH3-27, (IIIа)); 13C NMR (CDCl3; the δ values, ppm, were obtained through the С/Н correlations in the HSQC and НМВС spectra): 173.8 (C-1), 134.1 (C-9), 129.6 (CH-5), 129.0 (CH-6), 126.05 (CH-10), 60.2 (–OCOСН2CH3), 39.4 (CH2-8), 39.0 (CH2-24, (IIa)), 36.6 (CH2-23, (IIIa)), 34.4 (CH-24, (IIIa)), 33.7 (CH2-2), 30.0–28.5 (CH2-pool), 29.4 (CH2-25, (IIIa)), 28.65 (CH2-12), 28.5 (CH2-11), 28.0 (CH-25, (IIa)), 26.6 (CH2-4), 25.25 (CH2-7), 24.85 (CH2-3), 22.6 (CH3-26, 27, (IIa)), 19.2 (CH3-27, (IIIa)), 14.2 (−OCOСН2CH3), and 11.3 (CH3-26, (IIIa)); MS of the ethyl ester of (5Z,9Z)-9-chloro-25-methyl-5,9-hexacosadienoic acid (IIа) (EI, 70 eV), m/z (Irel, %): 468 [M]+ (0.04), 432/433 [M – HCl/M – Cl]+ (16/14), 422/424 [M – EtOH]+ (2/0.7), 417 [M – HCl – CH3]+ (0.7), 405/407 [M ‒ EtO ‒ H2O]+ (0.7/0.2), 387 [M ‒ Cl ‒ EtOH]+ (16), 369 [M ‒ HCl – EtO ‒ H2O]+ (5), 345 [M ‒ 88 ‒ Cl]+ (6), 312/314 (6/2), 276 (3), 154/155 (51/45), 109 (61), 95 (28), 88 (24), 81 (100), 67 (54), and 55/57 (36/34); MS of the ethyl ester of (5Z,9Z)-9-chloro-24-methyl-5,9-hexacosadienoic acid (IIIа) (EI, 70 эВ), m/z (Irel, %): 468 [M]+ (0.02), 432/433 [M – HCl/M – Cl]+ (16/14), 422/424 [M – EtOH]+ (2/0.7), 405/407 [M ‒ EtO ‒ H2O]+ (0.7/0.2), 403 [M – HCl – CH3]+ (0.8), 387 [M ‒ Cl ‒ EtOH]+ (16), 369 [M ‒ HCl – EtO ‒ H2O]+ (6), 345 [M ‒ 88 ‒ Cl]+ (6), 312/314 (5/1.7), 276 (4), 154/155 (52/44), 109 (61), 95 (29), 88 (24), 81 (100), 67 (56), and 55/57 (39/49).
Preparation of FA derivatives. Pyrrolidine derivatives (pyrrolidides) were synthesized by the treatment of FAs with N,O-bis(trimethylsilyl)trifluoroacetamide (Alfa Aesar, USA) and pyrrolidine (Aldrich, Germany) at room temperature [47]. Before analysis, the reaction mixture was concentrated in a vacuum. Free FAs were ethylated with N-nitroso-N-ethyl urea (Sigma, Germany). To obtain dimethyl disulfide adducts, the FA ethyl esters were treated with dimethyl disulfide (Sigma-Aldrich, France) according to the method described by Christie [48]. However, after the reaction was stopped with an aqueous solution of Na2S2O3, the reaction mixture was extracted with hexane (5 × 0.5 mL), and the hexane extract was dried in a vacuum before analysis. 4,4-Dimethyloxazoline derivatives were prepared by the reaction with 2-amino-2-methyl-1-propanol in pyridine in the presence of sodium borohydride (all reagents from Sigma-Aldrich, Germany) [49]. Hydrogenation (3–5.5 h) was performed over an Adams catalyst in ethanol.
The mass spectra of the derivatives of new FA are given in Supplementary.
CONCLUSIONS
The structural analysis of fatty acids from a sponge of the Penares genus was performed for the first time using GC-MS and, in some cases, 1H and 13C NMR spectroscopy. We found 71 acids with chain lengths from C12 to C28 including 12 new compounds. Previously unknown (5Z,9Z)-9-chloro-5,9-dienoic acids represent the first examples of chlorinated demospongic acids. The features of the studied FA mixture are the high content of components with monomethylated chains (>50%) and the almost complete replacement of common demospongic acids with their chloro derivatives due to, presumably, the activity of chlorinating enzymes of microorganisms associated with the sponge. The data from the analysis of FAs from Penares sp. not only expand our knowledge about the diversity of biomolecules but also contribute to understanding the origin of the structural features of some secondary metabolites of the sponge genus.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
Dembitsky, V.M., Rezanka, T., and Srebnik, M., Chem. Phys. Lipids, 2003, vol. 123, pp. 117‒155. https://doi.org/10.1016/S0009-3084(03)00020-3
Rodkina, S.A., Russ. J. Mar. Biol., 2005, vol. 31, pp. S49–S60. https://doi.org/10.1007/s11179-006-0015-3
Bergé, J.-P. and Barnathan, G., Marine Biotechnology I. Advances in Biochemical Engineering/Biotechnology, Ulber, R. and Le Gal, Y., Eds., Berlin, Heidelberg: Springer, 2005, vol. 96, pp. 49–125. https://doi.org/10.1007/b135782
Řezanka, T. and Sigler, K., Prog. Lipid Res., 2009, vol. 48, pp. 206‒238. https://doi.org/10.1016/j.plipres.2009.03.003
Manjari Mishra, P., Sree, A., and Panda, P.K., Springer Handbook of Marine Biotechnology, Kim, S.K., Ed., Berlin, Heidelberg: Springer, 2015, pp. 851–868. https://doi.org/10.1007/978-3-642-53971-8_36
Kornprobst, J.-M. and Barnathan, G., Mar. Drugs, 2010, vol. 8, pp. 2569‒2577. https://doi.org/10.3390/md8102569
Dembitsky, V.M. and Srebnik, M., Prog. Lipid Res., 2002, vol. 41, pp. 315‒367. https://doi.org/10.1016/S0163-7827(02)00003-6
Hwang, B.S., Lee, K., Yang, C., Jeong, E.J., and Rho, J.-R., J. Nat. Prod., 2013, vol. 76, pp. 2355‒2359. https://doi.org/10.1021/np400793r
Lyakhova, E.G., Kolesnikova, S.A., Kalinovsky, A.I., Dmitrenok, P.S., Nam, N.H., and Stonik, V.A., Steroids, 2015, vol. 96, pp. 37–43. https://doi.org/10.1016/j.steroids.2015.01.009
Lyakhova, E.G., Kolesnikova, S.A., Kalinovsky, A.I., Afiyatullov, Sh.Sh., Dyshlovoy, S.A., Krasokhin, V.B., Minh, Ch.V., and Stonik, V.A., Tetrahedron Lett., 2012, vol. 53, pp. 6119‒6122. https://doi.org/10.1016/j.tetlet.2012.08.148
Kobayashi, J., Cheng, J.-F., Ishibashi, M., Wälchli, M.R., Yamamura, Sh., and Ohizumi, Y., J. Chem. Soc., Perkin Trans. 1, 1991, pp. 1135–1137. https://doi.org/10.1039/P19910001135
Alvi, Kh.A., Jaspars, M., Crews, Ph., Strulovici, B., and Oto, E., Bioorg. Med. Chem. Lett., 1994, vol. 4, pp. 2447‒2450. https://doi.org/10.1016/S0960-894X(01)80407-8
Nakao, Y., Maki, T., Matsunaga, Sh., van Soest, R.W.M., and Fusetani, N., J. Nat. Prod., 2004, vol. 67, pp. 1346‒ 1350. https://doi.org/10.1021/np049939e
Takada, K., Uehara, T., Nakao, Y., Matsunaga, Sh., van Soest, R.W.M., and Fusetani, N., J. Am. Chem. Soc., 2004, vol. 126, pp. 187‒193. https://doi.org/10.1021/ja037368r
Fujita, M., Nakao, Y., Matsunaga, Sh., Seiki, M., Itoh, Y., van Soest, R.W.M., and Fusetani, N., Tetrahedron, 2001, vol. 57, pp. 1229‒1234. https://doi.org/10.1016/S0040-4020(00)01128-5
Ushio-Sata, N., Matsunaga, Sh., Fusetani, N., Honda, K., and Yasumuro, K., Tetrahedron Lett., 1996, vol. 37, pp. 225‒228. https://doi.org/10.1016/0040-4039(95)02134-5
Ando, H., Ueoka, R., Okada, Sh., Fujita, T., Iwashita, T., Imai, T., Yokoyama, T., Matsumoto, Y., van Soest, R.W.M., and Matsunaga, Sh., J. Nat. Prod., 2010, vol. 73, pp. 1947‒ 1950. https://doi.org/10.1021/np1003565
Bergquist, P.R., Lawson, M.P., Lavis, A., and Cambie, R.C., Biochem. Syst. Ecol., 1984, vol. 12, pp. 63–84. https://doi.org/10.1016/0305-1978(84)90012-7
Lawson, M.P., Bergquist, P.R., and Cambie, R.C., Biochem. Syst. Ecol., 1984, vol. 12, pp. 375–393. https://doi.org/10.1016/0305-1978(84)90070-X
Budzikevich, G., Djerasi, K., and Williams, D., Interpretation of Mass Spectra of Organic Compounds, Wolfson N.S., Ed., Moscow: Mir. 1966, pp. 323.
The LipidWeb. Mass Spectrometry of Alkyl Esters. Ethyl Esters of Fatty Acids. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/others/alkesters/index.htm
The LipidWeb. Mass Spectra of Fatty Acid Alkyl Esters— Archive. Ethyl Esters of Fatty Acids. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/others/others-arch/index.htm
The LipidWeb. Mass Spectrometry of Fatty Acid Pyrrolidides. Dienoic Fatty Acids. Part 2. Conjugated and Bis- and Polymethylene-Interrupted Dienes. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/pyrrolidides/pyrrol-2db-2/index.htm
The LipidWeb. Mass Spectrometry of DMOX Derivatives. Dienoic Fatty Acids. Part 2. Conjugated and Bis- and Polymethylene-Interrupted Dienes. https://www.lipidmaps.org/resources/lipidweb/lipidweb_html/ms/dmox/dmox-2db-2/index.htm
The LipidWeb. Mass Spectrometry of Fatty Acid Pyrrolidides. Saturated Branched-Chain Fatty Acids. https://www.lipidmaps.org/resources/lipidweb/lipidweb_html/ms/pyrrolidides/pyrrol-sbr/index.htm
The LipidWeb. Pyrrolidine Derivatives of Fatty Acids. Archive of Mass Spectra. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/pyrrolidides/pyrrol-arch/index.htm
Santalova, E.A. and Denisenko, V.A., Nat. Prod. Commun., 2017, vol. 12, pp. 1913–1916. https://doi.org/10.1177/1934578X1701201225
Dérien, S., Klein, H., and Bruneau, Ch., Angew. Chem. Int. Ed. Engl., 2015, vol. 54, pp. 12112–12115. https://doi.org/10.1002/anie.201505144
Gunstone, F.D., Chem. Phys. Lipids, 1993, vol. 65, pp. 155–163. https://doi.org/10.1016/0009-3084(93)90049-9
Akasaka, K., Shichijyukari, S., Meguro, H., and Ohrui, H., Biosci. Biotechnol. Biochem., 2002, vol. 66, pp. 1719–1722. https://doi.org/10.1271/bbb.66.1719
Santalova, E.A., Denisenko, V.A., and Dmitrenok, P.S., Molecules, 2020, vol. 25, p. 6047. https://doi.org/10.3390/molecules25246047
Andersson, B.A., Prog. Chem. Fats Other Lipids, 1978, vol. 16, pp. 279–308. https://doi.org/10.1016/0079-6832(78)90048-4
Santalova, E.A. and Denisenko, V.A., Lipids, 2017, vol. 52, pp. 73–82. https://doi.org/10.1007/s11745-016-4214-1
Knothe, G., Lipids, 2006, vol. 41, pp. 393–396. https://doi.org/10.1007/s11745-006-5110-x
Zhang, J.Y., Yu, Q.T., and Huang, Z.H., J. Mass Spectrom. Soc. Japan, 1987, vol. 35, pp. 23–30. https://doi.org/10.5702/massspec.35.23
The LipidWeb. Mass Spectrometry of Dimethyloxazoline and Pyrrolidine Derivatives. Cyclic Fatty Acids. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/dmox/dmox-cyclic/index.htm
The LipidWeb. Mass Spectrometry of Methyl Esters. Saturated Branched-Chain Fatty Acids. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/methesters/me-0dbbr/index.htm
The LipidWeb. Unesterified (Free) Fatty Acids. https://www.lipidmaps.org/resources/lipidweb/index.php?page=lipids/simple/ffa/index.htm
Thiel, V., Jenisch, A., Wörheide, G., Löwenberg, A., Reitner, J., and Michaelis, W., Org. Geochem., 1999, vol. 30, pp. 1–14. https://doi.org/10.1016/S0146-6380(98)00200-9
The LipidWeb. Fatty Acids: Branched-Chain. https://www.lipidmaps.org/resources/lipidweb/index.php?page=lipids/fa-eic/fa-branc/index.htm
The LipidWeb. Fatty Acids: Natural Cyclic. https://www.lipidmaps.org/resources/lipidweb/index.php?page=lipids/fa-eic/fa-cycl/index.htm
Reiswig, H.M., Mar. Ecol., 1981, vol. 2, pp. 273–293. https://doi.org/10.1111/j.1439-0485.1981.tb00271.x
Hedrick, D.B., Peacock, A.D., Long, Ph., and White, D.C., Lipids, 2008, vol. 43, pp. 843–851. https://doi.org/10.1007/s11745-008-3206-1
Fejzagić, A.V., Gebauer, J., Huwa, N., and Classen, T., Molecules, 2019, vol. 24, p. 4008. https://doi.org/10.3390/molecules24214008
Bayer, K., Scheuermayer, M., Fieseler, L., and Hentschel, U., Mar. Biotechnol., 2013, vol. 15, pp. 63–72. https://doi.org/10.1007/s10126-012-9455-2
Wang, J., Pang, X., Chen, Ch., Gao, Ch., Zhou, X., Liu, Y., and Luo, X., Chin. J. Chem., 2022, vol. 40, pp. 1729–1750. https://doi.org/10.1002/cjoc.202200064
Vetter, W. and Walther, W., J. Chromatogr. А, 1990, vol. 513, pp. 405–407. https://doi.org/10.1016/S0021-9673(01)89466-8
The LipidWeb. Mass Spectrometry of Methyl Esters. Derivatization of Double Bonds in Fatty Acids for Structural Analysis. https://www.lipidmaps.org/resources/lipidweb/index.php?page=ms/methesters/me-dbderivs/index.htm
Santalova, E.A. and Svetashev, V.I., Nat. Prod. Commun., 2022, vol. 17, pp. 1–8. https://doi.org/10.1177/1934578X221131408
ACKNOWLEDGMENTS
The work was performed using the equipment of the Collective Facilities Center “The Far Eastern Center for Structural Molecular Research (NMR/MS) of G.B. Elyakov Pacific Institute of Bioorganic Chemistry of the Far Eastern Branch of Russian Academy of Sciences” (NMR spectroscopy and mass spectrometry).
The authors thank O.P. Moiseenko, Ph.D. L.P. Ponomarenko, and E.G. Lyakhova for their assistance in the GC-MS analysis and the isolation of the FA ethyl esters, academician V.A. Stonik for discussion of some issues of this work, Ph.D. V.A. Denisenko and V.V. Isakov and operators N.V. Zvyagintsev and D.V. Denisenko for recording the NMR spectra.
Funding
The work was supported by the Russia Foundation for Basic Research (grant no. 20-03-00014).
Author information
Authors and Affiliations
Contributions
All authors made equal contributions to the writing of the article.
Corresponding author
Ethics declarations
This article does not contain any studies involving patients or animals as test objects.
Informed consent was not required for this article. No conflict of interest was declared by the authors.
Additional information
Publisher's Note. Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations: FA, fatty acids; ECL, equivalent chain length; 1Н, 1Н-COSY, proton-proton correlation spectroscopy; НМВС, heteronuclear multiple bond correlation; HSQC, heteronuclear single-quantum coherence.
Supplementary information
Rights and permissions
About this article

Cite this article
Santalova, Е.А., Kolesnikova, S.А. 9-Chloro-5,9-dienoic and Other Fatty Acids from Marine Sponge Penares sp.. Russ J Bioorg Chem 50, 418–431 (2024). https://doi.org/10.1134/S1068162024020249
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162024020249