
Abstract
Here, we described a facile method for synthesis of novel 1-piperidinyl and 1-piperazinyl-3-thioxo-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile compounds as well as the previously synthesized 1-morpholinyl derivative by the reaction of 3-amino-1-thioxo-1,5,6,7-tetrahydro cyclopenta[c]thiopyran-4-carbonitrile with piperidine, piperazine and morpholine, respectively through Dimorth rearrangement. A series of new 1-amino-2-substituted-5-morpholinyl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-c]pyridine was synthesized by the reaction of the morpholinyl cyclopenta[c]pyridine thione with α-halogenated carbonyl compounds namely: ethyl chloroacetate, chloroacetone and ω-bromoacetophenone via two synthetic routes. The ethyl 1-amino-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carboxylate was utilized as a powerful precursor for the synthesis of novel heterocycles containing cyclopenta[d]thieno[2,3-c]pyridine moiety. Hydrazinolysis of the amino thienopyridine carboxylate ester with hydrazine hydrate 99% upon reflux under solvent-free conditions produced the corresponding amino-carbohydrazide, which was used as a versatile intermediate to prepare new heterocyclic systems. The chemical structures of all newly synthesized compounds were elucidated based on their elemental and spectral analyses. Furthermore, some of these compounds revealed promising anti-inflammatory activity using carrageenan-induced rat paw edema assay compared with indomethacin as a reference drug.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
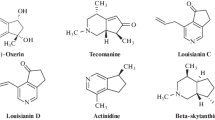
Pyridines and cyclopentapyridines are abundant scaffolds in nature and privileged compounds in medicinal and agricultural chemistry. Cyclopentapyridines are considered the major structure of many naturally occurring monoterpenoid alkaloids namely tecomanine [1], (+)-oxerin, actinidine [2] (-)-plectrodorine [3, 4], skytanthine [5, 6], as well as anti-cancer and anti-androgenic alkaloids louisianins A–D [7, 8]. Some cyclopenta[c]pyridine species revealed a significant affinity for the central nicotinic receptor [9] and also were found to be effective precursors for the synthesis of anticancer alkaloid camptothecin analogous [10]. Furthermore, thienopyridine compounds play an essential role in heterocyclic chemistry and display broad spectrum of therapeutic activities. Certain thieno[2,3-b]pyridines exhibit anti-inflammatory [11], antibacterial [12], antifungal [13], antiviral [14], anticancer [15], antihypertensive [16], antidepressant [17], neurotropic [18] and immune-stimulating activities [19].
On the other hand, thienopyrimidines are commonly utilized in a variety of applications in medicinal chemistry. Thienopyrimidine derivatives proved to possess several pharmaceutical activities as anti-inflammatory [20], antimicrobial [21], antitumor [22], anti-allergic [23], antioxidant [24], antiviral [25], antiplatelet [26] and antihypertensive properties [27].
In the light of the prior biological importance of cyclopentapyridine [28, 29], thienopyridine and thienopyrimidine, and in a resumption of our program for the synthesis of novel biologically active heterocycles involving thienocyclopentapyridine and thienotetrahydro- isoquinoline moieties [30–38]. In the current investigation, we have synthesized novel 1-piperidinyl and 1-piperazinyl-3-thioxo-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile compounds as well as a series of new cyclopenta[d]thieno[2,3-b]pyridines and cyclopenta[4'',5'']pyrido[3',2':4,5]thieno[3,2-d]-pyrimidines.
As demonstrated, non-steroidal anti-inflammatory drugs (NSAIDs) are the most important medicines used for reducing pain and swelling accompanied by inflammation. However, the long-term clinical usage of NSAIDs is associated with remarkable side effects including bleeding, gastrointestinal lesions and nephrotoxicity [39–41]. Therefore, one of the main objectives of our research is the synthesis of new heterocyclic compounds that could be used as new anti-inflammatory drugs. Hence, the assumed promising biological activities of the cyclopentapyridine and thienopyridine encouraged us to investigate the in vivo anti-inflammatory activities of cyclopentapyridothienopyrimidine heterocycles in comparison with the indomethacin standard drug. The results showed a promising activity to reduce the severity of foot edema caused by carrageenan.
RESULTS AND DISCUSSION
Based on the inspiring proficiency and continuous attempts to synthesize new heterocyclic compounds having promising biological activity, herein, we represented the synthesis of new heterocycles fused or attached to the cyclopentathienopyridine moiety, namely: pyrimidine, pyrazole, and pyrrole ring systems. The previously described method was used to synthesize 3-amino-1-thioxo-1,5,6,7-tetrahydrocyclopenta[c]thiopyran-4-carbonitrile (I) and its conversion into cyclopentathienopyridine derivatives via Dimroth rearrangement utilizing alkaline medium [30]. This transformation was reported in this paper employing various amines namely: morpholine, piperidine, and piperazine to give 1-(substituted)-3-thioxo-2,3,6,7-tetrahydro-5H-cyclopenta[c]pyridine-4-carbonitrile (IIa–c). Compound (IIa) was used as an important intermediate in the synthesis of several novel heterocyclic compounds containing thienocyclopentapyridine and cyclopentapyridothieno pyrimidine moieties (Scheme 1). IR spectrum of compound (IIb) revealed the appearance of absorption band at 3350 cm–1 specific for NH group and disappearance of NH2 band in the initial compound (I).1H NMR of (IIb) in DMSO-d6 exhibited singlet signal at 11.70 ppm unique for NH group. Furthermore, IR of (IIc) represented two absorption bands at 3440 and 3247 cm–1 typical for NH pyridine and NH piperazine. 1H NMR displayed two singlet signals at 6.69 and 11.76 ppm attributed to NH piperazine and NH pyridine, respectively (Scheme 1).

Scheme 1 . Dimorth rearrangement of the 3-amino cyclopenta[c]thiopyran-4-carbonitrile (I) to the 1-substituted cyclopenta[c]pyridine-3-thione compounds (IIa–c).
A sequence of 1-amino-2-substituted-5-(morpholin-4-yl)-7,8-dihydro-6H-cyclopenta[c]thieno-[2,3-b]pyridine derivatives (IVa–c) was synthesized by two distinct methodologies. The first methodology is represented by the reaction of 1-morpholinyl cyclopentapyridine thione (IIa) with various alkylating agents such as ethyl chloroacetate, chloroacetone, and phenacyl bromide in refluxing ethanol and fused sodium acetate to afford the sulfanyl derivatives (IIIa–c). Compounds (IIIa–c) underwent Thorpe-Ziegler cyclization upon treatment with ethanolic sodium ethoxide solution to afford the target bi-functionally cyclopenta[d]thieno[2,3-b]pyridines (IVa–c) in quantitative yields. Compounds (IVa–c) were directly obtained via an alternative methodology through the reaction of pyridinethione (IIa) with the described alkylating agents in ethanol and the presence of anhydrous potassium carbonate. The products obtained by the two pathways were identical in all features and their structures were elucidated by elemental and spectral analyses. FT-IR analysis of compound (IVa) indicated the absence of the CN group’s absorption band at 2205 cm–1 in compound (IIIa) and appearance of two bands at 3472, 3357 cm–1 characteristic to NH2 in addition to a band at 1660 cm–1 unique for α, β-unsaturated CO of ester group. 1H NMR displayed triplet and quartet signals at 1.37 and 4.31 ppm attributed to the ethoxyl group as well as singlet signal at δ 5.93 ppm distinctive for NH2 group instead of a signal at 3.92 ppm characteristic for SCH2 group in the starting material. 13C NMR exhibited a signal at δ 165.89 ppm particular for CO of the ester group. Also, the mass spectrum of the amino ester (IVa) showed a molecular ion peak and base peak at 347.19 (Scheme 2).

Scheme 2 . Synthesis of 1-amino-2-substituted-5-morpholinyl cyclopenta[d]thieno[2,3-b]pyridines (IVa–c) by two alternative methods.
The ethyl 1-amino-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carboxylate (IVa) was utilized as a multipurpose precursor for synthesis of other novel heterocyclic compounds containing cyclopenta[d]thieno[2,3-b]-pyridine ring system. Thus, hydrazinolysis of the ester group in compound (IVa) with hydrazine 99% under solvent-free conditions afforded the corresponding amino-carbohydrazide (V) in a moderate yield. Elucidation of the chemical structure of compound (V) was confirmed by elemental and spectral investigation. FT-IR exhibited absorption bands at 3472, 3357, 3336, and 3263 cm–1 specific for NH and 2NH2 groups accompanied with the disappearance of bands characteristic of the carbonyl ester group and appearance of absorption band at 1660 cm–1 typical for amidic CO group instead. 1H-NMR of the carbohydrazide (V) in DMSO-d6 showed two singlet signals at δ 4.33 and 6.56 ppm particular for 2NH2 and a singlet signal at 8.76 ppm attributed to NH group. Mass spectrum exhibited a molecular ion peak at 333.16. Additionally, reaction of the amino-ester (IVa) with 2,5-dimethoxytetrahydrofuran in acetic acid yielded the pyrrolyl ester derivative (VI). The structure of compound (VI) was proved by elemental and spectral analyses. FT-IR presented the disappearance of bands characteristic of NH2 group in compound (IVa). 1H-NMR spectrum in DMSO-d6 affirmed two multiplet signals at δ 6.23 and 6.87 ppm distinctive for 4CH groups of pyrrolyl protons. In an analogous manner, the reaction of the pyrrolyl ester (VI) with hydrazine hydrate afforded the pyrrolyl carbohydrazide (VII) in an excellent yield. When the amino-ester (IVa) was allowed to react with chloroacetyl chloride in dioxane at 70–80°C followed by neutralization with diluted sodium carbonate solution, the chloroacetyl amino derivative (VIII) was obtained. FT-IR analysis of compound (VIII) indicated the presence of absorption band at 3210 cm–1 particular for NH group as well as bands at 1708 and 1681 cm–1 attributed to CO ester and CO amide, respectively. 1H-NMR in DMSO-d6 emphasized singlet signal at δ 10.29 ppm particular for NH. 13C NMR revealed signal at 161.90 and 166.30 distinctive for CO ester and the amidic CO, respectively.
The suggested fragmentation pattern of the 1‑amino-2-acetyl-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[c]thieno[2,3-b]pyridine (IVb) is displayed as follows in Scheme 3.

Scheme 3 . Fragmentation pattern of compound (IVb).
Subsequently, the reaction of chloroacetyl amino (VIII) with potassium thiocyanate in ethanol afforded a new tetracyclic ring system namely: ethyl 2-((4-morpholin-4-yl-7-oxo-2,3,7,8-tetrahydro-1H-cyclopenta-[4',5']pyrido[3',2':4,5]thieno[3,2-d]pyrimidin-9-yl)-sulfanyl) acetate (IX) in one step. FT-IR spectrum showed an absorption band at 3435 cm–1 that is characteristic of NH as well as bands at 1739 and 1697 cm–1 unique to CO ester and CO pyrimidine, respectively. 1H NMR in DMSO-d6 displayed triplet and quartet signals at δ 1.20, 4.17 ppm specific for the ethyl ester as well as singlet signal at δ 4.14 ppm distinctive for SCH2 group. The cholroacetyl amino (VIII) underwent a nucleophilic substitution reaction with aniline to give the aryl aminoacetyl-thienopyridine derivative (X). The structure of compound (X) was elucidated by FT-IR and 1H-NMR spectra. FT-IR of compound (X) showed two absorption bands at 3441 and 3179 cm–1 unique for 2NH. 1H NMR in DMSO-d6 demonstrated a singlet signal at δ 3.92 ppm distinctive for CH2 and two singlet signals at δ 6.13 and 9.96 ppm particular for NH-Ph and CONH. 13C NMR of compound (X) revealed two signals at 162.16 characteristic for CO ester and at 171.35 ppm attributed to amidic CONH group (Scheme 4).
The proposed reaction mechanism for formation of the ethyl cyclopentapyridothienopyrimidinyl sulfanyl acetate (IX) from chloroacetylamino derivative (VIII) was suggested to proceed by nucleophilic substitution of the chlorine atom in compound (VIII) with the carbonitrile group to afford the non-isolated intermediate A. Next, nucleophilic addition of NH to the CN group yielded the imino thiazolidine intermediate B. Finally, formation of the target ethyl oxopyrimidine sulfanyl acetate compound (IX) was carried out by a nucleophilic addition of NH of thiazolidinone to the ester group accompanied with removing the ethoxyl group as an excellent leaving group, acting as a strong nucleophile to break the thiazolidinone as depicted in the following Scheme 5.

Scheme 4 . Reaction of amino ester (IVa) with hydrazine hydrate, 2,5-dimethoxytetrahydrofuran and chloroacetyl chloride affording the synthesized compounds (V), (VI) and (VIII).

Scheme 5 . Mechanism of formation the ethyl pyridothienopyrimidinyl mercapto acetate (IX) from chloroacetyl amino derivative (VIII).
Alternatively, the o-amino carbohydrazide compound (V) was utilized as a versatile precursor for the synthesis of novel cyclopenta[d]thieno[2,3-b]pyridine and cyclopenta[4'',5'']pyrido[3',2':4,5]thieno[3,2-d]pyrimidine heterocycles. Condensation of the carbohydrazide (V) with 1,3-dicarbonyl compounds namely: ethyl acetoacetate and acetylacetone afforded the corresponding pyrazolyl derivatives (XII), (XIII). Thus, the reaction of the amino carbohydrazide (V) with ethyl acetoacetate yielded the corresponding hydrazone (XI), which underwent heterocyclization to the pyrazolone derivative (XII) upon heating in acetic acid and catalytic drops of conc. sulfuric acid. Formation of compounds (XI) and (XII) was established by FT-IR and 1H NMR spectra. FT-IR of (XI) exhibited absorption band at 1728 cm–1 attributed to CO ester group. 1H NMR in CDCl3 revealed the presence of triplet and quartet signals at δ 1.29 and 4.21 ppm attributed to CH3 and CH2 of the ethoxy group. Consequently, the FT-IR spectrum of pyrazolone compound (XII) exhibited absorption band at 1644 cm–1 for CO group. 1H NMR in DMSO-d6 displayed singlet signal at δ 2.90 specific for CH2 pyrazole. Analogously, condensation of (V) with acetylacetone in ethanol afforded the dimethyl pyrazolyl derivative (XIII). FT-IR of (XIII) presented absorption bands at 3447 and 3342 cm–1 distinctive for NH2 group and at 1639 for CO group. 1H-NMR in DMSO-d6 showed two singlet signals at δ 1.85 and 2.02 ppm for 2CH3 groups attached to pyrazole and singlet signal at δ 6.29 endorsed to CH pyrazole.
The amino-carbohydrazide (V) was reacted with triethyl orthoformate in ethanol in the presence of catalytic drops of acetic acid to afford the ethoxymethyleneaminopyridothieno- pyrimidinone (XIV). The structure of compound (XIV) was confirmed by elemental and spectral data. FT-IR exhibited an absorption band at 1686 cm–1 specific for C=O. 1H-NMR in CDCl3 displayed triplet and quartet signals at δ 2.12, 3.36 ppm unique for the ethyl group as well as two singlet signals at δ 11.35 and 8.44 ppm characteristic for CH pyrimidine and CH=N groups, respectively.
Condensation of the carbohydrazide (V) with benzaldehyde in ethanol gave the corresponding Schiff’s base (XV). FT-IR spectrum of compound (XV) showed absorption bands at 3465, 3313, and 3139 cm–1 unique for NH, NH2 groups and at 1628 cm–1 specific for amidic CO group. 1H-NMR in DMSO-d6 emphasized singlet signal at 11.22 ppm distinctive for NH group as well as multiplet signals at δ 7.43–7.79 ppm characteristic for aromatic protons.
The arylidene hydrazone (XV) was cyclized using acetic anhydride or triethyl orthoformate to give the corresponding pyridothienopyrimidinone derivatives (XVI) and (XVII). FT-IR of the latter compounds exhibited disappearance of band characteristic for NH2 group. The 1H-NMR of compound (XVI) revealed singlet signals at δ 1.21 ppm specific for CH3 pyrimidine and at δ 8.98 ppm characteristic of CH benzylidene. Also, the 1H-NMR spectrum of compound (XVII) showed singlet signals at δ 8.62 and 9.27 ppm characteristic of CH benzylidene and CH pyrimidine (Scheme 6).
Biological Screening
Nonsteroidal anti-inflammatory medicines (NSAIDs) are a class of pharmaceuticals that have been approved by the FDA for use as antipyretic, anti-inflammatory, and analgesic agents. Because of these effects, NSAIDs can be used to treat muscle pain, dysmenorrhea, arthritic, pyrexia, gout, and migraines diseases as well as opioid-sparing medicines in some acute trauma instances. So, one of the essential targets of our research is a synthesis of novel heterocycles having predominant importance in biological and medicinal chemistry. As a result, the projected promising biological activities of cyclopentathienopyridines pressured us to study the anti-inflammatory significance of cyclopentathienopyridine heterocycles in comparison to standard pharmaceuticals.
In the present study, we focused on comparing the anti-inflammatory impacts between compounds having similar functional groups and carrying the same cyclopentathienopyridine moiety to effectively emphasize the structure-activity relationship (SAR) among them. The anti-inflammatory activity assessment for some of the newly synthesized cyclopentathienopyridine compounds was measured at 1, 2, 3, and 4 h after carrageenan injection. Indomethacin was used as authentic drug. The data which was listed in (Tables 1, 2 and Figs. 2, 3) indicated that all the tested compounds (IVa), (VIII), (XIII), (XIV) and (XV) revealed promising anti-inflammatory effects. It is worthy to be mentioned that compound (XIV) revealed the highest anti-inflammatory activity with paw edema inhibition very close to indomethacin during the period of the experiment (1–4 h). Compounds (VIII), (XIII), (XIV) and (XV) displayed rapid and significant activity, while (IVa) showed moderate effect comparable to indomethacin after 30 min. Compounds (VIII) and (XIV) exhibited the best effectiveness after 1 and 2 h of treatment. After 3 h, compounds (VIII), (XIII) and (XIV) revealed excellent performance with similar edema inhibition to the reference drug, whereas compounds (IVa) and (XV) presented moderate effects parallel to indomethacin. After 4 hrs, all the examined compounds revealed highly promising anti-inflammatory efficacy excluding compound (XV) comparable to indomethacin.

Scheme 6 . Synthesis of new pyrazolyl cyclopentathienopyridine compounds (XII), (XIII) and the novel tetracyclic cyclopentapyridothienopyrimidine heterocycles (XVI), (XVII).

Some naturally occurring monoterpenoid alkaloids containing cyclopentapyridine moiety.

The relationship of Paw edema inhibition (mm) with time (min).

Aplot of paw edema inhibition (%) with time (min).
Practically, carrageenan-induced paw edema was a prototype of the exudative phase of inflammation. The development of edema was described as biphasic, the initial phase is due to the release of mediators of inflammation like histamine, serotonin, dopamine and kinens in the first hour, however, the more pronounced second phase is related to prostaglandins release in 2–3 h. Consequently, the significant anti-inflammatory effect of compounds (IVa), (VIII), (XIII), (XIV) and (XV) may be due to the inhibitory effect exerted predominately on the mediators of inflammation [44–46].
EXPERIMENTAL
All melting points are uncorrected and measured on a Fisher–John apparatus. Elemental analyses were carried out at the Micro Analytical Center of Chemistry Department, Assiut University. Their results were found to be in good agreement (±0.2%) with the calculated values. FT-IR spectral analyses were recorded using potassium bromide disks on a FT-IR 820/PC Shimadzu. 1H NMR and 13C NMR spectra for compounds (IVa), (VII), (VIII), (IX), (X), (XI), (XIII), (XIV), (XV), (XVI) and (XVII) were obtained on Bruker (1H NMR: 400 MHz, 13C NMR 100 MHz) as well as Varian Mercury VX-300 NMR (1H NMR: 300MHz, 13C NMR 75 MHz) spectrometers for compounds (IIb), (IIc), (IIIa–c), (IVb), (IVc), (V), (VI) and (XII) in CDCl3 and DMSO-d6 using tetramethyl silane (TMS) as an internal standard (chemical shifts were expressed in ppm). All the reactions were monitored by thin layer chromatography (TLC) technique on silica gel coated on aluminum sheets (Silica Gel 60F254 Merck) using UV light. All reactions were carried out under an air atmosphere. Mass spectra were obtained on ISQ 7000 (70 eV) apparatus at Chemistry Department Lab, Faculty of Science, Assiut University. Using the computer Chem Draw professional 13.0, the structures of all produced compounds were drawn and named. Compound (IIa) was produced using a procedure reported in the literature [30].
Synthesis of compound (IIa,c). General procedure. A solution of the 3-amino-1-thioxo-1,5,6,7-tetrahydrocyclopenta[c]thiopyran-4-carbonitrile (I) (10.00 g, 0.05 mol) and different amines (0.05 mol) was refluxed under solvent-free conditions for 1.5 h or until H2S gas was ceased. The solid precipitate that formed on cooling, was filtered off, dried and recrystallized from the proper solvent.
1-(Piperidin-1-yl)-3-thioxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridine-4-carbonitrile (IIb). Produced as brown crystals in 64% (8.0 g) yield, mp 128–130°C. FT-IR: 3350 (NH), 2204 (CN), 2929, 2850 (CH aliphatic) and 1253 (C=S). 1H NMR (DMSO-d6): 1.68 (m, 6H, 3CH2: C3–C5 piperidinyl), 2.01 (m, 2H, CH2: C6 cyclopenteno), 2.84 (m, 2H, CH2: C7 cyclopenteno), 3.16 (m, 2H, CH2: C5 cyclopenteno), 3.52 (m, 4H, 2CH2: (CH2)2N: C2 and C6 piperidinyl), 11.70 (s, 1H, NH). Anal. Calcd. for C14H17N3S (259.37): C, 64.83; H, 6.61; N, 16.20; S, 12.36%. Found C, 64.78; H, 6.57; N, 16.16; S, 12.31%.
1-(Piperazin-1-yl)-3-thioxo-3,5,6,7-tetrahydro-2H-cyclopenta[c] pyridine-4-carbonitrile (IIc). Produced as pale brown crystals in 72% (9.00 g) yield, mp 160–162°C. FT-IR: 3440, 3247 (2NH) and 2206 (CN). 1H NMR (DMSO-d6): 1.88 (m, 6H, 3CH2: C5–C7 cyclopenteno), 2.71 (m, 4H, 2CH2: C3 and C5 (CH2)2NH piperazinyl), 3.41 (m, 4H, 2CH2: C2 and C6 (CH2)2N piperazinyl), 6.69 (s, 1H, NH piperazine), 11.76 (s, 1H, NH pyridine). Anal. Calcd. for C13H16N4S (260.36): C, 59.97; H, 6.19; N, 21.52; S, 12.31%. Found C, 59.93; H, 6.25; N, 21.47; S, 12.25%.
Synthesis of compounds (IIIa–c). General procedure. A mixture of compound (IIa) (3.00 g, 15 mmol), alkylating agent (e.g. ethyl chloroacetate, chloroacetone, and phenacyl bromide) (15 mmol) in ethanol (40 mL) and fused sodium acetate (3.00 g, 3.40 mmol) was refluxed for 1 hr. Then the reaction mixture was allowed to cool. The solid precipitate formed was filtered off, dried, and recrystallized from ethanol.
Ethyl 2-((4-cyano-1-morpholin-4-yl-6,7-dihydro-5H-cyclopenta[c]pyridin-3-yl) thio)acetate (IIIa). Produced as yellow crystals in 75% (3.00 g) yield, mp 120–122°C. FT-IR: 2981, 2964, 2850 (CH aliphatic), 2205 (CN) and 1748 (CO ester). 1H NMR (CDCl3): 1.27 (t, J = 7.00 Hz, 3H, CH3 ester), 2.12 (m, 2H, CH2: C6 cyclopenteno), 2.92 (m, 4H, 2CH2: C5 and C7 cyclopenteno), 3.61 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.79 (m, 4H, 2CH2: (CH2)2O morpholinyl), 3.92 (s, 2H, CH2CO) and 4.19 (q, J = 7.10 Hz, 2H, CH2 ester). Anal. Calcd. for C17H21N3O3S (347.43): C, 58.77; H, 6.09; N, 12.09; S, 9.23%. Found: C, 58.73; H, 6.14; N, 12.174; S, 9.19%.
Morpholin-4-yl-3-((2-oxopropyl)thio)-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile (IIIb). Produced as yellow crystals in 89% (3.20 g) yield, mp 140–142°C. FT-IR: 2965, 2944, 2850 (CH aliphatic), 2204 (CN) and 1717 (CO acetyl). 1H NMR (CDCl3): 2.12 (m, 2H, CH2: C6 cyclopenteno), 2.28 (s, 3H, COCH3) 2.92 (m, 4H, 2CH2: cyclopenteno), 3.58 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.77 (m, 4H, 2CH2: (CH2)2O morpholinyl) and 3.85 (s, 2H, CH2CO). Anal. Calcd. for C16H19N3O2S (317.41): C, 60.55; H, 6.03; N, 13.24; S, 10.10 Found: C, 60.49; H, 6.09; N, 13.30; S, 10.04%.
1-Morpholin-4-yl-3-((2-oxo-2-phenylethyl)thio)-6,7-dihydro-5H-cyclopenta[c]pyridine-4-carbonitrile (IIIc). Produced as yellow crystals in 95% (4.20 g) yield, mp 210–212°C. FT-IR: 2969, 2905, 2841 (CH aliphatic), 2200 (CN) and 1689 (CO benzoyl). 1H NMR (CDCl3): 2.11 (m, 2H, CH2: C6 cyclopenteno), 2.85 (m, 2H, CH2: C7 cyclopenteno), 2.93 (m, 2H, CH2: C5 cyclopenteno), 3.35 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.52 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.62 (s, 2H, CH2 benzoyl) and 7.50–8.06 (m, 5H, ArH). Anal. Calcd. for: C21H21N3O2S (379.48). C, 66.47; H, 5.58; N, 11.07; S, 8.45%. Found: C, 66.41; H, 5.52; N, 11.10; S, 8.50%.
Synthesis of compound (IVa–c). General procedure. Method a: To a solution of the appropriate 1-morpholin-4-yl-3-(alkylthio)-6,7-dihydro-5H-cyclopenta-[c]pyridin-4-carbonitrile (IIIa–c) (0.01 mol) in absolute ethanol (25 mL), few drops of sodium ethoxide solution (prepared from 0.50 g of divided clean sodium in ethanol (20 mL) was added and the mixture was refluxed for 15 min and then left to cool. The solid product was filtered off, dried, and recrystallized from ethanol. Method b: A mixture of compound (IIb) (2.75 g, 0.01 mol), alkylating agent (0.01 mol) and anhydrous potassium carbonate (3.00 g.0.02 mol) in ethanol (40 mL) was refluxed for 3 h, then was allowed to cool. The solid precipitate was filtered off, dried, and recrystallized from ethanol.
Ethyl 1-amino-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carboxylate (IVa). Produced as white crystals in 91% (2.50 g) yield, mp 160–162°C. FT-IR: 3472, 3357 (NH2), 2958, 2917, 2896 (CH aliphatic) and 1660 (α,β-unsat. CO). 1H NMR (CDCl3): 1.37 (t, J = 7.20 Hz, 3H, CH3 ester), 2.21 (m, 2H, CH2: C7 cyclopenteno), 2.9 (m, 2H, CH2: C6 cyclopenteno), 3.24 (m, 2H, CH2: C8 cyclopenteno), 3.49 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.82 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.31 (q, J = 7.20 Hz, 2H, CH2 ester) and 5.93 (s, 2H, NH2). 13C NMR (DMSO-d6): 14.57, 25.48, 31.45, 31.99, 48.12, 60.13, 66.97, 117.37, 124.08, 148.30, 150.32, 158.35, 159.03, 165.89 ppm. EI-MS (m/z): 347.19 [M+]. Anal. Calcd. for C17H21N3O3S (347.43): C, 58.77; H, 6.09; N, 12.09; S, 9.23%. Found: C, 58.73; H, 6.05; N, 12.14; S, 9.18%.
1-(1-Amino-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridin-2-yl)ethan-1-one (IVb). Produced as green crystals in 80% (2.20 g) yield, mp 168–170°C. FT-IR: 3435, 3290 (NH2), 2959, 2849 (CH aliphatic), 1574 (COCH3) cm–1. 1H NMR (CDCl3): 2.21 (m, 2H, CH2: C7 cyclopenteno), 2.38 (s, 3H, CH3 acetyl), 2.90 (m, 2H, CH2: C6 cyclopenteno), 3.20 (m, 2H, CH2: C8 cyclopenteno), 3.52 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.82 (m, 4H, 2CH2: (CH2)2O morpholinyl) and 6.75 (s, 2H, NH2) ppm. 13C NMR (75 MHz, DMSO-d6): 25.34, 28.89, 32.10, 47.98, 66.92, 104.79, 116.88, 124.08, 149.04, 151.05, 159.11, 192.28. EI-MS (m/z): 317.18 [M+]. Anal. Calcd. for C16H19N3O2S (317.41): C, 60.55; H, 6.03; N, 13.24; S, 10.10%. Found: C, 60.49; H, 6.08; N, 13.31; S, 10.03%.
(1-Amino-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridin-2-yl)(phenyl)methanone (IVc). Produced as pale green crystals in 95% (2.60 g) yield, mp 210–212°C. FT-IR: 3508, 3327 (NH2), 2959, 2913, 2840 (CH aliphatic), 1575 (COPh). 1H NMR (CDCl3): 2.23 (m, 2H, CH2: C7 cyclopenteno), 2.92 (m, 2H, CH2: C6 cyclopenteno), 3.27 (m, 2H, CH2: C8 cyclopenteno), 3.54 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.82 (m, 4H, 2CH2: (CH2)2O morpholinyl), 7.15–7.54 (m, 5H, ArH) and 7.87 (s, 2H, NH2). EI-MS (m/z): 379.17 [M+]. Anal. Calcd. for C21H21N3O2S (379.48): C, 66.47; H, 5.58; N, 11.07; S, 8.45%. Found: C, 66.40; H, 5.65; N, 11.02; S, 8.39%.
1-Amino-5-(morpholin-4-yl)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carbohydrazide (V). The amino ester (IVa) (0.50 g, 1.40 mmol) and hydrazine hydrate 99% (2 mL, 0.06 mol) were fused under solvent free conditions for 30 min then absolute ethanol (7 mL) was added dropwise. The reaction mixture was refluxed for additional 2 h. The solid product formed during reflux was recrystallized from ethanol to give pale yellow crystals in 52% (0.25 g) yield, mp 264–266°C. FT-IR: 3472, 3357, 3336, 3263 (NH, 2NH2), 2957, 2921, 2849 (CH aliphatic), 1660 (CONH). 1H-NMR (DMSO-d6): 2.07 (m, 2H, CH2: C7 cyclopenteno), 2.84 (m, 2H, CH2: C6 cyclopenteno), 3.24 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.69 (m, 4H, 2CH2: (CH2)2O morpholinyl), 3.69 (m, 2H, CH2: C8 cyclopenteno),4.33 (s, 2H, NH2 carbohydrazide), 6.56 (s, 2H, NH2 thieno), 8.76 (s, 1H, CONH) ppm. EI-MS (m/z): 333.16 [M+]. Anal. Calcd. for: C15H19N5O2S (333.41): C, 54.04; H, 5.74; N, 21.03; S, 9.62%. Found: C, 54.10; H, 5.69; N, 21.10; S, 9.58.
Ethyl-5-morpholin-4-yl-1-(1H-pyrrolyl)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carboxylate (VI). A mixture of amino ester compound (IVa) (0.50 g, 1.40 mmol) and (0.2 mL, 1.50 mmol) 2,5-dimethoxytetrahydrofuran in acetic acid (2 mL) was gently refluxed for 1h. The produced compound which formed on cooling was filtered off, dried, and recrystallized from ethanol as white crystals in 70% (0.40 g) yield, mp 170–172°C. FT-IR: 2964, 2984, 2920, 2893 (CH aliphatic) and 1685 (CO ester). 1H NMR (DMSO-d6): 1.09 (t, 3H, J = 7.10 Hz, CH3 ester), 1.89 (m, 2H, CH2: C7 cyclopenteno), 2.27 (m, 2H, CH2: C6 cyclopenteno), 2.87 (m, 2H, CH2: C8 cyclopenteno), 3.46 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.72 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.11 (q, 2H, J = 7.10 Hz, CH2 ester), 6.23 (m, 2H, 2CH: C3, C4 pyrrolyl) and 6.87 (m, 2H, 2CH: C2, C5 pyrrolyl). Anal. Calcd. for C21H23N3O3S (397.49): C, 63.47; H, 5.84; N, 10.57; S, 8.07%. Found: C, 63.43; H, 5.90; N, 10.49; S, 8.02%.
5-Morpholin-4-yl-1-(1H-pyrrolyl)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carbohydrazide (VII). A mixture of the pyrrolyl ester (VI) (0.50 g, 1.30 mmol) and hydrazine hydrate 99% (0.60 mL, 0.015 mol) was refluxed in ethanol for 3 h. The produced compound which formed while hot during reflux was filtered off, dried, and recrystallized from ethanol–dioxane (2 : 1) mixture as white crystals in 83% (0.4 g) yield, mp 230–232°C. FT-IR: 3374, 3319, 3116 (NH2, NH), 2961, 2920, 2848 (CH aliphatic) and 1645 (CO amide). 1H NMR (DMSO-d6): 2.09 (m, 2H, CH2: C2 cyclopenteno), 2.87 (m, 2H, CH2: C3 cyclopenteno), 3.44 (m, 2H, CH2: C1 cyclopenteno), 3.50 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.74 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.29 (s, 2H, NH2), 6.30 (m, 2H, 2CH: C3'',C4'' pyrrolyl), 6.94 (m, 2H, 2CH: C2'',C5'' pyrrolyl) and 7.73 (s, 1H, CONH). 13C NMR (DMSO-d6): 25.47, 29.42, 32.25, 48.31, 66.58, 110.32, 122.25, 123.75, 126.47, 127.15, 130.80, 150.95, 155.34, 157.68, 160.87. Anal. Calcd. for C19H21N5O2S (383.47): C, 59.51; H, 5.52; N, 18.26; S, 8.36%. Found: C, 59.46; H, 5.45; N, 18.33; S, 8.31%.
Ethyl 1-(2-chloroacetamido)-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carboxylate (VIII). A suspension of the amino-ester compound (IVa) (0.50 g, 1.40 mmol) and chloroacetyl choride (0.8 mL, 0.01 mol) in dioxane (20 mL) was heated on water bath at 70–80°C for 2 h. Then the reaction mixture was allowed to cool and neutralized with diluted sodium carbonate solution (10%) till just alkaline. The solid product was collected and recrystallized from ethanol as white crystals in 83% (0.50 g) yield; mp 138–140°C. FT-IR: 3210 (NH), 2959, 2836 (CH aliphatic), 1708 (CO unsat. ester), 1681 (CO amide). 1H NMR (DMSO-d6) δ (ppm): 1.31 (t, 3H, J = 7.20 Hz, CH3 ester), 2.06 (m, 2H, CH2: C7 cyclopenteno), 2.92 (m, 2H, CH2: C6 cyclopenteno), 3.11 (m, 2H, CH2: C8 cyclopenteno), 3.47 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.73 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.28 (s, 2H, CH2Cl), 4.38 (q, 2H, J = 7.20 Hz, CH2 ester) and 10.29 (s, 1H, NH). 13C NMR (DMSO-d6): 14.57, 25.42, 31.21, 32.34, 43.08, 48.13, 61.48, 66.59, 119.37, 122.07, 126.06, 133.82, 152.20, 156.84, 158.13, 161.90, 166.30. Anal. Calcd. for C19H22ClN3O4S (423.91): C, 53.83; H, 5.23; Cl, 8.36; N, 9.91; S, 7.56%. Found: C, 53.76; H, 5.28; Cl, 8.32; N, 9.85; S, 7.60%.
Ethyl 2-((4-morpholin-4-yl-7-oxo-2 ,3,7,8-tetrahydro-1H-cyclopenta[4'',5'']pyrido[3',2':4,5]thieno-[3,2-d]pyrimidin-9-yl)thio)acetate (IX). A mixture of the chloro acetamido (VIII) (0.40 g, 1.00 mmol) and slightly excess of potassium thiocynate (0.12 g, 1.25 mmol) in ethanol (5 mL) was refluxed for 2 h. The solid product was collected and recrystallized from ethanol–dioxane mixture (3 : 1) as white crystals in 75% (0.30 g) yield, mp > 360°C. FT-IR: 3435 (NH), 2917, 2848 (CH aliphatic), 1739 (C=O unsat ester), 1697 (CONH). 1H NMR (DMSO-d6): 1.20 (t, 3H, J = 7.10 Hz, CH3 ester), 2.12 (m, 2H, CH2: C2 cyclopenteno), 2.99 (m, 2H, CH2: C3 cyclopenteno), 3.38 (m, 2H, CH2: C1 cyclopenteno), 3.52 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.73 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.14 (s, 2H, CH2CO), 4.17 (q, 2H, J = 7.10 Hz, CH2 ester), 13.02 (s, 1H, NH pyrimidinone). Anal. Calcd. for C20H22N4O4S2 (446.54): C, 53.68; H, 4.95; N, 12.55; S, 14.36%. Found: C, 53.61; H, 4.88; N, 12.51; S, 14.41%.
Ethyl 5-morpholin-4-yl-1-(2-(phenylamino)acetamido)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]-pyridine-2-carboxylate (X). The chloro acetamido derivative (VIII) (0.50 g, 1.20 mmol) and aniline (1.00 mL, 2.74 mmol) were refluxed in ethanol (10 mL) for 2 h. The solid product which formed upon cooling was filtered off, dried and recrystallized from ethanol as white crystals in 75% (0.45 g) yield, mp 190–192°C. FT-IR: 3441, 3179 (2NH), 3043 (CH aromatic), 2959, 2947, 2848 (CH aliphatic), 1696 (C=O ester), 1663 (CO amidic). 1H NMR (DMSO-d6): 1.28 (t, 3H, J = 7.00 Hz, CH3 ester), 1.97 (m, 2H, CH2: C7 cyclopenteno), 2.88 (m, 2H, CH2: C6 cyclopenteno), 3.00 (m, 2H, CH2: C8 cyclopenteno), 3.46 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.73 (m, 4H, 2CH2: (CH2)2O morpholinyl), 3.92 (s, 2H, COCH2), 4.24 (q, 2H, J = 7.10 Hz, CH2 ester), 6.13 (s, 1H, NHPh), 6.63–7.13 (m, 5H, ArH) and 9.96 (s, 1H, NHCO). 13C NMR (DMSO-d6): 14.66, 25.46, 31.42, 32.30, 47.76, 48.12, 61.38, 66.58, 113.10, 117.22, 117.62, 122.19, 129.27, (134.73, 148.70, 152.51, 156.08, 158.08, 162.16, 171.35. Anal. Calcd. for C25H28N4O4S (480.58): C, 62.48; H, 5.87; N, 11.66; S, 6.67%. Found: C, 62.53; H, 5.91; N, 11.61; S, 6.60%.
Ethyl-3-(2-(1-amino-5-morpholino-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carbonyl)-hydrazono)butanoate (XI). A solution of the carbohydrazide (V) (1.00 g, 3.00 mmol) and ethyl acetoacetate (0.6 mL, 4.60 mmol) was refluxed in ethanol (10 mL). The solid precipitate which separated out during reflux was collected and recrystallized from ethanol as yellow crystals in 71% (0.95 g) yield, mp 218–220°C. FT-IR: 3493, 3339 (NH2), 3137 (NH), 2961, 2849 (CH aliphatic), 1728 (C=O ester), 1646 (C=O amide). 1H-NMR (CDCl3): 1.29 (t, 3H, J = 6.80 Hz, CH3 ester), 1.91 (s, 3H, CH3C=N), 2.10 (m, 2H, CH2: C7 cyclopenteno), 2.88 (m, 2H, CH2: C6 cyclopenteno), 3.24 (m, 2H, CH2: C8 cyclopenteno), 3.39 (s, 2H, CH2), 3.49 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.75 (m, 4H, 2CH2: (CH2)2O morpholinyl), 4.21 (q, 2H, J = 6.80 Hz, CH2 ester), 6.52 (s, 2H, NH2), 8.70 (s, 1H, CONH). Anal. Calcd. for C21H27N5O4S (445.54): C, 56.61; H, 6.11; N, 15.72; S, 7.20%. Found: C, 56.56; H, 6.05; N, 15.65; S, 7.14%.
2-(1-Amino-5-(morpholin-4-yl)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridine-2-carbonyl)-5-methyl-2,4-dihydro-3H-pyrazol-3-one (XII). A mixture of the butanoate ester (XI) (0.50 g, 1.10 mmol) and acetic acid (2 mL) was refluxed for 1 h. The solid precipitate which formed on hot during reflux was filtered off, dried and recrystallized from ethanol as yellow crystals in 89% (0.40 g) yield, mp 218–220°C. FT-IR: 3473, 3353 (NH2), 2932, 2965 (CH aliphatic), 1644 (C=O). 1H-NMR (DMSO-d6):1.91 (s, 3H, CH3), 2.11 (m, 2H, CH2: C7 cyclopenteno), 2.51 (m, 2H, CH2: C6 cyclopenteno), 2.90 (s, 2H, CH2 pyrazole), 3.27 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.45 (m, 2H, CH2: C8 cyclopenteno), 3.72 (m, 4H, 2CH2: (CH2)2O morpholinyl), and 6.67 (s, 2H, NH2) ppm. Anal. Calcd. for C19H21N5O3S (399.47): C, 57.13; H, 5.30; N, 17.53; S, 8.03%. Found: C, 57.19; H, 5.24; N, 17.59; S, 8.09%.
(1-Amino-5-(morpholin-4-yl)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridin-2-yl)(3,5-dimethyl-1H-pyrazol-1-yl)methanone (XIII). The carbohydrazide (V) (0.40 g, 1.20 mmol) and acetylacetone (0.70 mL, 7.0 mmol) were fused in absence of solvent for 10 min., then ethanol was added, and the reaction mixture was refluxed for 2 h. The solid precipitate which separated out while hot during reflux was collected and recrystallized from ethanol as yellow crystals in 83% (0.40 g) yield, mp 258–260°C. FT-IR: 3447, 3342 (NH2), 2915, 2848 (CH aliphatic), 1639 (C=O). 1H-NMR (DMSO-d6): 1.85 (s, 3H, CH3: C5 pyrazolyl), 2.02 (m, 3H, CH3: C3 pyrazolyl), 2.02 (m, 2H, CH2: C7 cyclopenteno), 2.84 (m, 4H, 2CH2: C6 and C8 cyclopenteno), 3.26 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.70 (m, 4H, 2CH2: (CH2)2O morpholinyl), 6.29 (s, 1H, CH pyrazole), and 7.07 (s, 2H, NH2). Anal. Calcd. for: C20H23N5O2S (397.50): C, 60.43; H, 5.83; N, 17.62; S, 8.07%. Found: C, 60.47; H, 5.78; N, 17.56; S, 8.02%.
8-Ethoxymethylene-amino-4-(morpholin-4-yl)-7-oxo-1,2,3,7-tetrahydro-8H-cyclopenta[4'',5'']pyrido-[3',2':4,5]thieno[3,2-d]pyrimidin-7(8H)-one (XIV). A suspension of the carbohydrazide (V) (0.50 g, 1.5 mmol) and triethyl orthoformate (2.00 mL, 13.50 mmol) was fused for 10 min., then few drops of acetic acid were added. The solid precipitate that produced on hot during reflux was recrystallized from ethanol as yellow crystals in 92% (0.55 g) yield, mp 288–290°C. FT-IR: 2986, 2857 (CH aliphatic), 1686 (C=O pyrimidine). 1H-NMR (DMSO-d6): 2.12 (t, 3H, J = 7.00 Hz, CH3 ester), 2.12 (m, 2H, CH2: C2 cyclopenteno), 2.51 (m, 2H, CH2: C3 cyclopenteno), 3.00 (m, 2H, CH2: C1 cyclopenteno) 3.36 (q, J = 7.00 Hz, 2H, CH2 ester), 3.56 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.75 (m, 4H, 2CH2: (CH2)2O morpholinyl), 8.44 (s, 1H, CHOEt) and 11.35 (s, 1H, CH pyrimidine). Anal. Calcd. for C19H21N5O3S (399.47): C, 57.13; H, 5.30; N, 17.53; S, 8.03%. Found: C, 57.20; H, 5.36; N, 17.48 S, 8.08%.
N '-Benzylidene-(1-amino-5-(morpholin-4-yl)-7,8-dihydro-6H-cyclopenta[d]thieno[2,3-b]pyridin-2-yl)-carbohydrazide (XV). A mixture of the carbohydrazide (V) (2.00 g, 6.0 mmol) and benzaldehyde (1.0 mL, 9.40 mmol) in ethanol (10 mL) was refluxed for 1 h. The solid product formed on hot during reflux was collected and recrystallized from dioxane to give canary yellow crystals in 92% (2.30 g) yield, mp 270–272°C. FT-IR: 3465, 3313, 3139 (NH, NH2), 1628 (C=O amide), 1570 (C=N). 1H-NMR (DMSO-d6): 2.10 (m, 2H, CH2: C7 cyclopenteno), 2.87 (m, 2H, CH2: C6 cyclopenteno), 3.29 (m, 2H, CH2: C8 cyclopenteno), 3.57 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.73 (m, 4H, 2CH2: (CH2)2O morpholinyl), 7.18 (s, 2H, NH2), 7.43–7.79 (m, 5H, ArH), 8.08 (s, 1H, CH benzylidene), 11.22 (s, 1H, CONH). Anal. Calcd. for C22H23N5O2S (421.52): C, 62.69; H, 5.50; N, 16.61; S, 7.61%. Found: C, 62.73; H, 5.54; N, 16.57; S, 7.65%.
8-(Benzylideneamino)-9-methyl-4-(morpholin-4-yl)-2,3-dihydro-1H-cyclopenta[4'',5'']pyrido[3',2':4,5]-thieno[3,2-d]pyrimidin-7(8H)-one (XVI). A sample of benzylidine carbohydrazide (XV) (0.20 g, 0.48 mmol) and acetic anhydride (1.50 mL, 0.015 mol) was refluxed for 2 h. The solid product which precipitated on hot during reflux was filtered off, dried and recrystallized from dioxane as yellow powder in 75% (0.15 g) yield, mp 208–210°C. FT-IR: 2951, 2917, 2837 (CH aliphatic), 1666 (CO pyrimidine), 1585 (C=N). 1H-NMR (DMSO-d6): 1.21 (s, 3H, CH3), 2.13 (m, 2H, CH2: C2 cyclopenteno), 2.96 (m, 2H, CH2: C3 cyclopenteno), 3.28 (m, 2H, CH2: C1 cyclopenteno), 3.52 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.74 (m, 4H, 2CH2: (CH2)2O morpholinyl), 7.59–7.96 (m, 5H, ArH), 8.98 (s, 1H, CH benzylidene) ppm. Anal. Calcd. for C24H23N5O2S (445.54): C, 64.70; H, 5.20; N, 15.72; S, 7.20%. Found: C, 64.65; H, 5.15; N, 15.68; S, 7.15%.
8-(Benzylideneamino)-4-(morpholin-4-yl)-2,3-dihydro-1H-cyclopenta[4'',5'']pyrido[3',2':4,5]thieno-[3,2-d]pyrimidin-7(8H)-one (XVII). A mixture of benzylidene amino (XV) (0.50 g, 1.19 mmol) and triethyl orthoformate (2 mL, 0.014 mol) in presence of few drops of glacial acetic acid (0.3 mL) was refluxed for 15 min. The solid product which produced on hot during reflux was filtered off, dried and recrystallized from dioxane as white crystals in 78% (0.40 g) yield, mp 288–290°C. FT-IR: 2961, 2895, 2864 (CH aliphatic), 1672 (CO pyrimidine), 1578 (C=N). 1H‑NMR (DMSO-d6): 2.13 (m, 2H, CH2: C2 cyclopenteno), 2.95 (m, 2H, CH2: C3 cyclopenteno), 3.30 (m, 2H, CH2: C1 cyclopenteno), 3.52 (m, 4H, 2CH2: (CH2)2N morpholinyl), 3.73 (m, 4H, 2CH2: (CH2)2O morpholinyl), 7.58–7.93 (m, 5H, ArH), 8.62 (s, 1H, CH benzylidene) and 9.27 (s, 1H, CH pyrimidine). Anal. Calcd. for C23H21N5O2S (431.51): C, 64.09; H, 4.91; N, 16.23; S, 7.43%. Found: C, 64.15; H, 4.85; N, 16.30; S, 7.47%.
In Vivo Anti-Inflammatory Assay
Anti-inflammatory activity for the newly synthesized compounds (IVa), (VIII), (XIII), (XIV) and (XV) were measured in vivo using a carrageenan-induced rat paw edema assay in comparison with indomethacin as a reference drug [44, 45]. The test is based on the pedal inflammation in rat paw induced by sub plantar injection of 100 µL of 1% freshly prepared solution of carrageenan in distilled water into the right-hind paws of each rat for all the groups; the tested compounds were dissolved in distilled water with sonication. Male adult albino rats (150–200 g) were divided into six groups; each group contains three animals. The thickness of the rat paw edema was measured by a Vernier Caliper (SMIEC, China). Animals of groups A/B/C were treated with a single dose of the tested compound, and group D was treated with the Indomethacin drug, respectively. Paw thickness was measured just before the carrageenan injection, that is, at “0 h” and then at 30 min 1, 2, 3, 4, and 5 h after carrageenan injection. Increasing in paw thickness was measured as a difference in the paw thickness at “0 h” and paw thickness at respective hours. The edema was expressed as a mean reduction in paw volume (mL) after treatment with tested compounds. The percent edema inhibition was calculated from the mean effect in the control and treated animals according to the following equation:

where: Vt, means an increase in paw volume of test; Vc, means an increase in paw volume of control group of rats.
Statistical Analysis
The results were analyzed by one-way analysis of variance (ANOVA) followed by Newman-Keuls multiple comparison test as a post-test. These analyses were carried out using a computer prism program for windows, version 3.0 (Graph pad software, Inc., San Diego, CA, US). The significant differences between groups were accepted at P < 0.05*, 0.01** or 0.001***, and the data are expressed as a mean ± standard error (SE).
CONCLUSIONS
In the current study, we have provided an easy access for synthesis of novel 1-amino-2-substituted-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno-[2,3-b]pyridines (IVa–c). Ethyl 1-amino-5-morpholin-4-yl-7,8-dihydro-6H-cyclopenta[d]thieno-[2,3-b]pyridine-2-carboxylate (IVa) was used as a versatile precursor for synthesis of new heterocyclic ring systems attached or fused to the cyclopenta-[d]thieno[2,3-b]pyridine moiety namely: pyrrole, pyrazole and pyrimidine. Most of the examined novel cyclopentathienopyridines exhibited promising anti-inflammatory activities compared to indomethacin. Based on the gained results, we can conclude that most of the examined compounds can be considered as potential anti-inflammatory drugs.
REFERENCES
Jones, G., Fales H.M., and Wildman, W.C., Tetrahedron Lett., 1963, vol. 4, pp. 397–400. https://doi.org/10.1016/S0040-4039(01)90645-8
Sakan, T., Fujino, A., and Murai, F., Bull. Chem. Soc., 1959, vol. 32, pp. 315–316.
Ohba, M., Izuta, R., and Shimizu, E., Tetrahedron Lett., 2000, vol. 41, pp. 10251–10255. https://doi.org/10.1016/S0040-4039(00)01824-4
Ohba, M., Izuta, R., and Shimizu, E., Chem. Pharm. Bull., 2006, vol. 54, pp. 63–67. https://doi.org/10.1248/cpb.54.63
Oppolzer, W. and Jacobsen, E. J., Tetrahedron Lett., 1986, vol. 27, pp. 1141–1144. https://doi.org/10.1016/S0040-4039(00)84199-4
Djerassi, C., Kutney, J.P., and Shamma, M., Tetrahedron, 1962, vol. 18, pp. 183–188. https://doi.org/10.1016/0040-4020(62)80038-6
Takamatsu, S., Kim, Y.P., Hayashi, M., Furuhata, K., Takayanagi, H., Komiyama, K., Woodruff, H.B., and Omura, S., J. Antibiot. (Tokyo), 1995, vol. 48, pp. 1090–1094. https://doi.org/10.7164/antibiotics.48.1090
Sunazuka, T., Zhi-Ming, T., Harigaya, Y., Takamatsu, S., Hayashi, M., Komiyama, K., and Omura, S., J. Antibiot. (Tokyo), 1997, vol. 50, pp. 274–275.https://doi.org/10.7164/antibiotics.50.274
Guandalini, L., Dei, S., Gualtieri, F., Romanelli, M. N., Scapecchi, S., Teodori, E., and Varani, K., Helv. Chim. Acta, 2002, vol. 85, pp. 96–107. https://doi.org/10.1002/1522-2675(200201)85:1
Gilbert, L., Patrick, H., Alain, P., Atassi, G., John, H., Bailly, C., Cimetiere, B., Leonce, S., and Laine, W., 2002, US Patent Appl. no. 2002/0077325.
Madhusudana, K., Shireesha, B., Naidu, V.G.M., Ramakrishna, S., Narsaiah, B., Rao, A.R., and Diwan, P.V., Eur. J. Pharmacol., 2012, vol. 678, pp. 48–54. https://doi.org/10.1016/j.ejphar.2011.12.019
Srivastava, B.K., Solanki, M., Mishra, B., Soni, R., Jayadev, S., Valani, D., Jain, M., and Patel, P.R., Bioorg. Med. Chem. Lett., 2007, vol. 17, pp. 1924–1929. https://doi.org/10.1016/j.bmcl.2007.01.038
Sangshetti, J.N., Khan, F.A.K., Chouthe, R.S., Damale, M.G., Shinde, D.B., Chin. Chem. Lett., 2014, vol. 25, pp. 1033–1038. https://doi.org/10.1016/j.cclet.2014.04.003
Schnte, M.E., Cudahy, M.M., and Scott, A., PCT Int. Appl. WO 0053610; Chem. Abstr., 2000, vol. 133, p. 222607g.
Munchhof, M.J., Soboloujaynes, S.B., Marx, M.A., US Patent 6492383; Chem. Abstr., 2003, vol. 138, p. 24721.
Adachi, I. and Hiramatsu, Y., Jpn. Patent no. 0352890; Chem. Abstr., 1991, vol. 115, p. 71573.
Kokai, T.K., Jpn. Patent no. 0616557; Chem. Abstr., 1994, vol., p. 290120.
Oganisyan, A.K., Noravyan, A.S., Dzhagatspanyan, I.A., and Melikyan, G.G., Pharm. Chem. J., 2003, vol. 37, p. 13–14.
Ooe, T., Sano, M., Kobayashi, H., and Kudome, M., Jpn. Kokai Tokkyo Koho JP. 0753562. Chem. Abstr., 1995, p. 256681k.
Alagarsamy, V., Meena, S., Ramseshu, K.V., Solomon, V.R., Thirumurugan, K., Dhanabal, K., and Murugan, M., Eur. J. Med. Chem. 2006, vol. 41, pp. 1293–1300. https://doi.org/10.1016/j.ejmech.2006.06.005
Litvinov, V.P., Russ. Chem. Bull., 2004, vol. 53, pp. 487–516.
Sasaki, S., Cho, N., Nara, Y., Harada, M., Endo, S., Suzuki, N., Furuya, S., and Fujino, M., J. Med. Chem., 2003, vol. 46, pp. 113–124. https://doi.org/10.1021/jm020180i
Gillespie, E., Dungan, K.W., Gomoll, A.W., and Seidehamel, R.J., Int. J. Immunopharmacol., 1985, vol. 7, pp. 655–660. https://doi.org/10.1016/0192-0561(85)90149-3
Kotaiah, Y., Nagaraju, K., Harikrishna, N., Venkata, R.C., Yamini, L., and Vijjulatha, M., Eur. J. Med. Chem., 2014, vol. 75, pp. 195–202. https://doi.org/10.1016/j.ejmech.2014.01.006
Rashad, A.E., Shamroukh, A.H., Abdel Megeid, R.E., Mostafa, A., El-Shesheny, R., Kandeil, A., Ali, M.A., and Banert, K., Eur. J. Med. Chem. 2010, vol. 45, pp. 5251–5257. https://doi.org/10.1016/j.ejmech.2010.08.044
Bruno, O., Brullo, C., Ranise, A., Schenone, S., Bondavalli, F., Barocelli, E., Ballabeni, V., Chiavarini, M., Tognolini, M., and Impicciatore, M., Bioorg. Med. Chem. Lett., 2001, vol. 11, pp. 1397–1400. https://doi.org/10.1016/S0960-894X(01)00221-9
Kim, Y., Kim, M., Park, M., Tae, J., Baek Park, K.D., and Choo, H., Molecules, 2015, vol. 20, pp. 5074–5084. https://doi.org/10.3390/molecules20035074
Paronikyan, E.G., Noravyan, A.S., Dashyan, Sh.Sh., and Minasyan, N.S., Chem. J. Arm., 2012, vol. 65, pp. 326–331.
Paronikyan, E.G., Dashyan, Sh.Sh., Minasyan, N.S., Stepanyan, G.M., and Babaev, E.V., Chem. Heterocycl. Compd., 2016, vol. 52, pp. 337–345. https://doi.org/10.1007/s10593-016-1887-6
Zaki, R.M., Kamal El-Dean, A.M., Radwan, Sh.M., and Ammar, M.A., Russ. J. Bioorg. Chem., 2020, vol. 46, pp. 85–96. https://doi.org/10.1134/S1068162020010148
Zaki, R.M., Kamal El-Dean A.M., Radwan, Sh.M., Alshammari, M.B., and Sayed, A.S.A., Arab. J. Chem., 2021, vol. 14, p. 103318. https://doi.org/10.1016/j.arabjc.2021.103318
Zaki, R.M., Kamal El-Dean A.M., Radwan, Sh.M., and Sayed, A.S.A., ACS Omega, 2020, vol. 5, pp. 252–264. https://doi.org/10.1021/acsomega.9b02604
Zaki, R.M., Radwan, Sh.M., and Kamal El-Dean, A.M., J. Chin. Chem. Soc., 2017, vol. 64, pp. 1417–1431. https://doi.org/10.1002/jccs.201700232
Zaki, R.M., Kamal El Dean, A.M., Abd El Monem, M.I., Seddik, M.A., Heterocycl. Comm., 2016, vol. 22, pp. 103–109. https://doi.org/10.1515/hc-2015-0204
Zaki, R.M., Kamal El-Dean, A.M., and Radwan, Sh.M., Afindad, 2011, vol. LXVIII, pp. 424–434.
Kamal El-Dean, A.M., Radwan, Sh.M., and Zaki, R.M., J. Chem. Res., 2010, vol. 34, pp. 596–602. https://doi.org/10.3184/030823410X12868184096970
Kamal El-Dean, A.M., Radwan, Sh.M., and Zaki, R.M., Eur. J. Med. Chem., 2011, vol. 46, pp. 567–578. https://doi.org/10.1016/j.ejmech.2010.11.036
Kamal El-Dean, A.M., Radwan, Sh.M., Zaki, R.M., J. Chin. Chem. Soc., 2008, vol. 55, pp. 1290–1299. https://doi.org/10.1002/jccs.200800193
Fiorucci, S., Santucci, L., and Distrutti, E., Dig. Liver Dis., 2007, vol. 39, pp. 1043–1450. https://doi.org/10.1016/j.dld.2007.09.001
Van Ryn, J., Trummlitz, G., and Pairet, M., Curr. Med. Chem., 2000, vol. 7, pp. 1145–1161. https://doi.org/10.2174/0929867003374255
Whittle, B.J.R., Eur. J. Pharmacol., 2004, vol. 500, pp. 427–439. https://doi.org/10.1016/j.ejphar.2004.07.042
Winter, C.A., Risley, E.A., and Nuss, G.W., Proc. Soc. Exp. Biol. Med., 1962, vol. 111, pp. 544–547. https://doi.org/10.3181/00379727-111-27849
Adeyemi, O.O., Okpo, S.O., and Ogunti, O.O., Fitoterapia, 2002, vol. 73, pp. 375–380.
Brooks, P.M. and Day, R.O., New. Engl. J. Med., 1991, vol. 324, pp. 1716–1725. https://doi.org/10.1056/NEJM199106133242407
Vane, J. and Botting, R., FASEB J., 1987, vol. 1, pp. 89–96.
Nagalakshmi, G., Ind. J. Pharm. Sci., 2008, vol. 70, pp. 49–55. https://doi.org/10.4103/0250-474X.40331
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
This article does not contain any studies involving human or animals participants performed by any of the authors.
Supplementary Information
Rights and permissions
About this article

Cite this article
Zaki, R.M., Kamal El-Dean, A.M., Radwan, S.M. et al. Efficient Synthesis, Reactions and Anti-Inflammatory Evaluation of Novel Cyclopenta[d]thieno[2,3-b]pyridines and Their Related Heterocycles. Russ J Bioorg Chem 48 (Suppl 1), S121–S135 (2022). https://doi.org/10.1134/S1068162023010314
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162023010314