
Abstract
6-(Benzo[d]thiazol-2-yl)-4-oxo-(4H)-chromene-3-carbaldehyde was utilized to construct a novel series of fused chromone bearing benzothiazole moiety, namely chromeno[2,3-b]azetol, benzo[c]chromene, and chromeno[2,3-c]pyrazole derivatives, as well as, non-fused chromones, such as thiazolidinone, pyrazolone, and pyridine derivatives. The in vitro antitumor activities of the synthesized products against six cancer cell lines, including A594, HCT-116, MCF-7, HepPG2, PC3, and HFB4, were evaluated. Some compounds exhibited significant anticancer activities against both lung and colon cancer cells very close to that of a standard drug doxorubicin.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Cancer is considered one of the deadliest diseases worldwide. Unfortunately, till now there is no drug with 100-% efficiency against cancer due to problems of toxicity and drug resistance that accompany chemotherapeutics in cancer treatment [1, 2]. Thus, unremitting efforts have been devoted to discover effective medicaments. A large number of synthetic heterocyclic compounds have been investigated for treatment of cancer. In particular, natural and synthetic chromenes, which exhibited diverse pharmacological activities, have been utilized as anticancer agents [3–14]. In addition, benzothiazoles possess a wide range of biological activities [15, 16], particularly, anticancer activity [17–28]. In view of these findings, we report herein synthesis of 6-(benzothiazol-2-yl)-4-oxo-4H-chromene derivatives and evaluation of their anticancer potential against diverse human cancer cell lines, including lung A549, colon HCT-116, breast MCF-7, prostate PC3, liver HepPG2, and human normal melanocyte HFB4 cancer cell lines.
RESULTS AND DISCUSSION
Chemistry
3-Formylchromones are important precursors in the field of heterocyclic synthesis due to their wide range of applications as chemotherapeutics, in particular, as anticancer agents. Conventionally, 3-formylchromones are prepared by application of Vilsmeier-Haack reaction on the substituted o-hydroxyacetophenones. Heating phenyl 4-(benzo[d]thiazol-2-yl)acetate (I) with anhydrous aluminum chloride gave o-hydroxyacetophenone (II), which was readily transformed into the desired 3-formylchromone (III) following the aforementioned reaction type.
The literature survey revealed the importance of thiazolidinones [17, 29–31] and pyrazolones [32–36] as antitumor agents. Accordingly, treatment of an ethanolic solution of 3-formylchromone (III) with 2,4‑dioxothiazolidine or 3-methyl-N-phenylpyrazol-5-one in the presence of a catalytic amount of either anhydrous sodium acetate or piperidine afforded the condensation products, thiazolidinone and pyrazolone derivatives (IV) and (V), respectively (Scheme 1). The structures of these products were deduced on the basis of the disappearance of stretching frequency of oxoformyl functionality and appearance of strong absorptions at 1614 and 1612 cm–1, of the vinylic bond stretching absorptions, respectively, in the IR spectrum. Similarly, refluxing an ethanolic solution of compound (III) with ethyl cyanoacetate in the presence of few drops of piperidine provided ethyl 3‑(6-(benzo-[d]thiazol-2-yl)-4-oxo-4H-chromen-3-yl)-2-cyanoacrylate (VI) as a sole product. Higher δ values at 8.86, 10.44, and 9.19 ppm in the 1H NMR spectra of compounds (IV)–(VI), respectively, revealed the presence of vinylic CH proton. Presumably, the higher δ value in compound (IV) is mainly due to high desheilding effects exerted by both anisotropic effects of the two CO groups (both ring moieties) and the electron withdrawing effect of S–CO– of the thiazole moiety. However, the higher δ values of similar protons are influenced by the anisotropic fields of the two CO group and the electron withdrawing effects of either C=N group of pyrazole moiety in compound (V) or C≡N functionality in compound (VI).

Scheme 1 . Synthetic pathway of compounds (III)–(IX).
The product of the reaction of compound (III) with acetylacetone depends on the reaction conditions. Thus, submitting 3-formylchromone (III) to react with acetylacetone in acetic anhydride containing fused sodium acetate yielded the expected condensation product (VII). The structure of the latter compound was confirmed by the two bands at 1766 and 1662 cm–1 in the IR spectrum due to the coupling absorption bands of the two symmetrical carbonyl groups of diacetyl groups bonded to the same olefinic C=C double bond. On the other hand, when the reaction was carried out in ethanol-ammonium acetate, chromeno[2,3-b]azetol (VIII) has been obtained. Formation of compound (VIII) could be explained via the elimination of the incorporated acetylacetone segment with consequent annulation of azete ring as illustrated in Scheme 2.

Scheme 2 . Suggested mechanism for the formation of compound (VIII).
This speculation was supported by the synthesis of compound (VIII) from 3-vinylchromene derivative (VII) under the effect of ammonium acetate in boiling ethanol (Scheme 1). The structure of product (VIII) was deduced from IR spectrum, which revealed lack of the carbonyl absorption of chromone moiety; besides, it displayed two bands at 3441 and 1606 cm–1 referring to the presence of OH and C=N groups, respectively. 1H NMR spectrum of compound (VIII) exhibited a singlet signal at δ 10.19 ppm due to –OH group proton. Moreover, the lack of absorption signals owing to methyl group protons in the upfield region refers to the elimination of acetylacetone segment. Furthermore, mass spectrum of product (VIII) showed the molecular ion peak at m/z 306. Alternatively, acid catalyzed condensation of 3-formylchromone (III) with acetylacetone afforded dihydroxybenzo[c]chromene derivative (IX).
In continuation of this work, 3-formylchromone (III) was allowed to react with some primary aromatic and heterylamines, including, p-aminoacetophenone, methyl p-aminobenzoate, and/or 4-aminopyridine to give the corresponding Schiff bases (Xa), (Xb), and(XI), respectively (Scheme 3). It was assumed that (Xa) and (Xb) have been provided by condensation of the given substrate with one mole of the utilized amine to give the corresponding Schiff bases followed by 1,2‑addition of a second amine molecule in position 2 of chromenone moiety [37]. This assumption has been supported by the increase of the percentage yield of the latter products upon increase of the concentration of the utilized amine. Herein, when the reaction was carried out using equimolar ratios of both the substrate and the co-reactant amine, the percentage yields of (Xa) and (Xb) were 43 and 45%, respectively. In turn, when the concentration of the amine was double the ratio of the substrate (III), this improved the percentage yields of these products to be 75 and 78%, respectively.
Thiosemicarbazones are of considerable interest because of their chemistry and potential antitumor activity [38, 39]. Accordingly, reaction of ethanolic solution of 3-formylchromone (III) with thiosemicarbazide afforded chromone-3-thiosemicarbazone (XII) in a good yield (Scheme 3). The structure of the latter product was deduced on the basis of IR spectrum, which lacked the oxoformyl group absorption frequency, whereas it exhibited a broad and a coupling absorption bands at 3212 and 3133 cm–1 due to NH2 and NH groups. In addition, the 1H NMR spectrum revealed two singlets at δ 8.20 and 11.56 ppm with integration of two and one protons corresponding to the two latter groups, respectively.

Scheme 3 . Synthetic pathway of compounds (X)–(XIV).
It has been reported that some 1,3-thiazole and thiosemicarbazone compounds not only exhibited cytotoxicity in cancer cells, but also were able to cause irreversible cancer cell damage by inducing necrosis and apoptosis [40–42]. These findings prompted us to carry out a remarkable easy access to thiazolidin-4-one derivative (XIII). This target has been achieved by prolonged heating of thiosemicarbazone (XII) with chloroacetyl chloride in the presence of fused sodium acetate (Scheme 3). IR spectrum of compound (XIII) revealed a stretching frequency of oxo-group of thiazolone ring at 1701 cm–1. 1H NMR spectrum of the latter grouping exhibited two broad signals at δ 13.50 and 11.11 ppm, both integrate to one proton, and a relatively low absorption value, presumably, due to lactam–lactim NHCO \( \rightleftarrows \) N=COH tautomerzation. In addition, the mass spectrum showed peaks at m/z = 422 and 420 due to (M + 2]+) and molecular ion peaks, respectively.
Among heterocyclic cores, pyrano[2,3-c]pyrazoles are popular with pharmacological importance. Particularly, they showed reasonable cytotoxic activity in vitro against cancer cell lines, for instance, A549, compared with doxorubicin, a well-known anticancer drug [43]. Recently, based on the beneficial role of Schiff bases as good intermediates for the synthesis of pyrazoles, together with the synthesis of 3-styrylpyrazoles via cleavage of pyran ring upon treatment of 3-styrylchromones with hydrazine hydrate [44], a series of successful chemical transformations via hydrazinolysis of compounds (III), (VI), Schiff bases (Xa, b), and thiosemicarazone (XII) into the novel chromeno[2,3-c]pyrazole (XIV) has been accomplished. First, refluxing ethanolic solutions of the former four compounds (III), (VI), and/or Schiff bases (Xa, b) with the bidentate nucleophile, hydrazine hydrate, resulted in the synthesis of the latter desired compound (XIV) in moderate yields (Scheme 3). Reaction of (Xa) and/or (Xb) with hydrazine hydrate gave a mixture of methyl p-aminobenzoate and/or p-aminoacetophenone (recrystallized from light petroleum) and (XIV) (crystallized from benzene), respectively.
Alternatively, the latter product has been provided readily either by conducting a mixture of compound (III) and thiosemicarbazide with hydrazine hydrate in boiling DMF or via prolonged heating of thiosemicarbazone (XII) in DMF. Careful investigation of these products obtained from these alternative routes showed full identity of TLC, mp, mixed mp, and IR spectra with that formerly provided in the first experiment.
The structure of (XIV) was deduced from its spectral data. Thus, IR spectrum revealed the absence of the stretching frequency of pyrone oxo-group and revealed absorption bands at 3406 and 1605 cm–1 due to OH and C=N groups. The 1H NMR spectrum of (XIV) displayed one singlet integrated for one proton at 10.18 ppm owing to OH group, together with a multiplet due to 8H representing the seven aromatic proton and HC-3 of pyrazole ring at δ 7.94–6.92 ppm. Further, the mass spectrum of the assigned product showed the molecular ion peak at m/z 319. Chemical evidence for the structure of (XIV) was forthcoming from the fact that it gives positive reaction with FeCl3 test indicating the presence of OH-phenolic group. Proposed mechanisms for the synthesis of (XIV) from compounds (III) and/or thiosemicarbazone (XII) were formulated as depicted in Scheme 4; conversions of compounds (VI) and (Xa, b) into the same former compound was visualized in Scheme 5.
Antitumor Activity
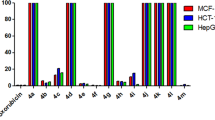
Cytotoxicity and in vitro anticancer evaluation. The in vitro antitumor activities of the synthesized compounds against six cancer cell lines and the results from Table 1 revealed that compounds (XIII) and (XIV) displayed the most promising antitumor activities against the A594 (lung cancer cells) and HCT-116 (colon carcinoma cells) cancer cell lines with IC50 values very close to those of doxorubicin, i.e. 5.00 ± 0.81 and 4.65 ± 0.86 µg/mL (versus 3.90 ± 0.42 µg/mL for doxorubicin) in case of lung cancer and 5.08 ± 0.80 and 4.90 ± 0.64 µg/mL (versus 4.20 ± 0.60 µg/mL for doxorubicin) in case of colon cancer. Compounds (X10a) and (XII) revealed moderate activity compared with the reference drug doxorubicin with IC50 values 12.10 ± 1.61 and 10.37 ± 1.82 µg/mL (versus 3.90 ± 0.42 µg/mL for doxorubicin) in case of lung cancer and 13.11 ± 2.48 and 7.07 ± 0.82 µg/mL (versus 4.20 ± 0.60 µg/mL for doxorubicin) in case of colon cancer. On the other hand, within the series of the compounds, it appeared that the tested compounds exerted very weak anticancer activity against liver (HepG2) and prostate (PC3) cancer cells, while in case of breast cancer (MCF-7) only compound (XIII) exerted moderate activity. The foregoing results also revealed that nearly all the tested compound showed no activities against the growth of normal HFB4 cells.
Structure–activity relationships.The cytotoxicity values of prepared compounds were matched to their structures and the following structure–activity relationships were proposed (Fig. 1):

Structure–activity relationship of the most potent anticancer compounds among those studied.
(1) Compound (XIII) has high potential activity against A594 and HCT-116 cancer cell lines and moderate activity against MCF-7, which may be attributed to the thiazolidenone ring with carbonyl group, which enhanced lipophilicity of the compound.
(2) Compound (XIV) has activities against the A594 and HCT-116 cancer cell lines closely potent to those of the control drug doxorubicin, which may be explained by the presence of fused pyrazole ring.
(3) Conversion of 3-formylchromone derivative (III) into thiosemicarbazide derivative (XII) enhances the antitumor activity, which may be due to presence of thiosemicarbazide group.
(4) Conversion of 3-formylchromone derivative (III) into Schiff base (Xa) increases the antitumor activity.

Scheme 4 . Proposed mechanisms for the formation of compound (XIV) from compounds (III) and (XII).

Scheme 5 . Suggested mechanisms for the transformation of compound (VI) and Schiff bases (Xa, b) to compound (XIV).
EXPERIMENTAL
Chemistry
All melting points were taken on Griffin and Geory melting point apparatus and are uncorrected. IR spectra (KBr; ν, cm–1) were recorded on Pye Unicam SP 1200 spectrophotometer using KBr Wafer technique. 1H NMR spectra were determined on a Varian Gemini 300 MHz spectrometer in DMSO-d6 using TMS as internal standard (chemical shifts in δ scale). 13C NMR spectra were measured on a Bruker Spectrometer PABBO 400 BB 100 MHz. EI-MS were measured on a Schimadzu-GC-MS, QP 1000 EX instrument operating at 70 eV. The homogeneity of the synthesized compounds was controlled by TLC using TLC aluminum sheets silica gel F254 (Merck).
1-(5-(Benzo[d]thiazol-2-yl)-2-hydroxyphenyl)ethanone(II). A mixture of 4-(benzo[d]thiazol-2-yl)phenyl acetate (I) (2.302 g, 0.008 mol) and anhydrous aluminum chloride (1.707 g, 0.012 mol) was heated in oil bath at 160°C for 6 h. The reaction mixture was cooled, and then poured onto ice/HCl with stirring. The precipitated solid was filtered off, washed successively with water, and recrystallized from light petroleum (bp 40–60°C) to give compound (II): yellow crystals; yield 60%, mp 169–170°C. IR spectrum: 3423 (OH), 3056 (CH aromatic), 2998 (CH aliphatic), 1645 (C=O). 1H NMR spectrum: 12.05 (s, 1H, OH, exchangeable with D2O), 8.45 (d, 1H, benzothiazol-H, J = 2.4 Hz), 8.11–8.21 (m, 2H, Ar-H), 8.04 (d, 1H, benzene-H, J = 8.1 Hz), 7.44, 7.54 (2t, 2H, benzothiazol-H, J = 7.2 Hz), 7.16 (d, 1H, benzene-H, J = 8.7 Hz), 2.74 (s, 3H, CH3). MS (m/z, %): 269 (M+•, 74), 254 (31), 227 (45), 134 (12), 108 (63), 43 (100). Calcd. for C15H11NO2S (269.312): C, 66.90; H, 4.11; N, 5.22, S; 11.91. Found: C, 66.84; H, 4.00; N, 5.09, S; 11.83.
6-(Benzo[d]thiazol-2-yl)-4-oxo-4H-chromene-3-carbaldehyde (III). Phosphorus oxychloride (2 mL, 0.02 mol) was added dropwise with continuous stirring to precooled DMF (10 mL), the mixture was further stirred at room temperature for 10 min and solution of o-hydroxyacetophenone derivative (II) (2.69 g, 0.01 mol) in DMF (10 mL) was added dropwise with continuous stirring. The reaction mixture was stirred at room temperature for further 2 h and left overnight, then poured onto crushed ice.The obtained solid was filtered off, dried, and recrystallized from ethanol to give 3-formylchromone (III): yellow crystals, yield 50%, mp 238–240°C. IR spectrum : 3063 (CH aromatic), 1699 (C=Oaldehyde), 1660 (C=Ochromone). 1H NMR spectrum: 10.14 (s, 1H, CHO), 8.99 (s, 1H, C2-H chromone), 8.54 (d, 1H, benzothiazol-H, J = 4.8 Hz), 8.22 (d, 1H, benzothiazol-H, J = 6 Hz), 7.97 (m, 2H, chromone-H), 7.52 (d, 1H, chromone-H, J = 5.4 Hz), 6.94, 7.40 (2t, 2H, benzothiazol-H, J = 5.4 Hz). MS (m/z, %): 307 (M+•, 13), 279 (M+-CO, 100), 253 (20), 251 (6),197 (13). Calcd. for C17H9NO3S (307.313): C, 66.44; H, 2.95; N, 4.56, S; 10.43. Found: C, 66.58; H, 2.82; N, 4.50, S; 10.55.
5-{[6-(Benzo[d]thiazol-2-yl)-4-oxo-4H-chromen-3-yl]methylene}thiazolidine-2,4-dione (IV). A mixture of 3-formylchromone (III) (1 g, 0.003 mol) and 2,4-dioxothiazolidine (0.38 g, 0.003 mol) in glacial acetic acid (20 mL) in presence of anhydrous sodium acetate (0.4 g, 0.003 mol) was refluxed for 2 h. The reaction mixture was then poured onto ice cooled water. The precipitated solid was filtered off, dried, and recrystallized from ethanol to give thiazolidindione derivative (IV): yellow crystals, yield 64%, mp > 300°C. IR spectrum: 3210 (NH), 3027 (CH aromatic), 1741, 1690 (2C=Othiazolidindion), 1657 (C=Ochromone), 1614 (C=C). 1H: 12.46 (s, 1H, NH, exchangeable with D2O), 8.86 (s, 1H, =C–H), 8.55 (d, 1H, benzothiazol-H, J = 2.4 Hz), 8.52 (d, 1H, benzothiazol-H, J = 2.1 Hz) , 8.19 (d, 1H, benzothiazol-H, J = 7.8 Hz), 8.12 (d, 1H, benzothiazol-H, J = 8.4 Hz), 7.92 (d, 1H, chromone-H, J = 9 Hz), 7.61–7.48 (m, 3H, Ar-H). MS (m/z, %): 406 (M+•, 54), 336 (23), 335 (100), 279(23), 253 (20), 225 (13), 197 (27). Calcd. for C20H10N2O4S2 (406.430): C, 59.10; H, 2.48; N, 6.89, S; 15.78. Found: C, 59.23; H, 2.34; N, 6.99, S; 15.66.
4-{[6-(Benzo[d]thiazol-2-yl)-4-oxo-4H-chromen-3-yl]methylene}-3-methyl-1-phenyl-1H-pyrazol-5(4H)-one (V). A solution of 3-formylchromone (III) (1 g, 0.003 mol) and 3-methyl-N-phenylpyrazol-5-one (0.54 g, 0.003 mol) in absolute ethanol (20 mL) in the presence of catalytic amount of piperidine was stirred at room temperature for 30 min. The precipitated solid was filtered off, washed with a little amount of ethanol, and recrystallized from benzene to give pyrazolone derivative (V): orange crystals, yield 90%, mp 260°C (decomp.). IR spectrum: 3048 (CH aromatic), 2995 (CH aliphatic), 1676 (C=Opyrazolone), 1630 (C=Ochromone), 1612 (C=C). 1H NMR spectrum: 10.44 (s, 1H, =C–H), 8.09 (d, 1H, benzothiazol-H, J = 2.7 Hz), 7.94 (d, 1H, benzothiazol-H, J = 2.7 Hz), 7.89–7.83 (m, 5H, Ar-H), 7.47–7.42 (m, 5H, Ar-H), 7.21 (s, 1H, chromone-H), 2.30 (s, 3H, CH3). MS (m/z, %): 463 (M+, 70), 435 (74), 435 (74), 434 (47), 406 (33), 329 (8), 253 (10), 197 (23). Calcd. for C27H17N3O3S (463.437): C, 69.98; H, 3.69; N, 9.07, S; 6.92. Found: C, 67.06; H, 3.79; N, 9.15, S; 6.80.
Ethyl 3-(6-(benzo[d]thiazol-2-yl)-4-oxo-4H-chromen- 3-yl)-2-cyanoacrylate (VI). A solution of 3-formylchromone (III) (1 g, 0.003 mol) and ethyl cyanoacetate (0.32 mL, 0.003 mol) in absolute ethanol (20 mL) in the presence of catalytic amount of piperidine was stirred at room temperature for 30 min. The precipitated solid was filtered off, washed with a little amount of ethanol, and recrystallized from benzene to give cyanoacrylate derivative (VI): brown crystals, yield 80%, mp 251–253°C. IR spectrum: 3071 (CH aromatic), 2982 (CH aliphatic), 2219 (CN), 1731 (C=Oester), 1660 (C=Ochromone). 1H NMR spectrum: 9.19 (s, 1H, =CH), 8.68 (d, 1H, benzothiazol-H, J = 9 Hz), 8.54 (d, 1H, benzothiazol-H, J = 9 Hz), 8.31 (s, 1H, C5-H chromone), 8.17, 8.12 (2d, 2H, C7 and C8-H chromone, J = 7.8 Hz), 7.94 (d, 1H, benzothiazol-H, J = 9 Hz), 7.61–7.48 (m, 2H, Ar-H), 4.34 (q, 2H, CH2, J = 7.2 Hz), 1.32 (t, 3H, CH3, J = 7.1 Hz). MS (m/z, %): 402 (M+•, 12), 329 (100), 253 (1), 197 (7). Calcd. for C22H14N2O4S (402.414): C, 65.66; H, 3.50; N, 6.96, S; 7.97. Found: C, 65.59; H, 3.62; N, 7.04, S; 8.05.
3-(2-Acetyl-3-oxobut-1-enyl)-6-(benzo[d]thiazol-2-yl)-4H-chromen-4-one (VII). A solution of 3-formylchromone (III) (1 g, 0.003 mol) and acetylacetone (0.328 mL, 0.003 mol) in freshly distilled acetic anhydride (10 mL) containing a catalytic amount of anhydrous sodium acetate was heated under reflux for 6 h. The reaction mixture was poured onto ice cold water and then extracted with ether. The ether layer was evaporated to give a residue, which was recrystallized from petroleum ether (bp 80–100°C) to afford chromone derivative (VII): yellow crystals, yield 60%, mp 138–140°C. IR spectrum: 3057 (CH aromatic), 2994 (CH aliphatic), 1766 (C=Oketone), 1662 (C=Ochromone). 1H NMR spectrum: 8.16 (s, 1H, CH=), 8.15–8.05 (m, 4H, Ar-H), 7.58–7.33 (m, 5H, Ar-H + C2-Hchromone) , 2.32 (s, 6H, 2 CH3). MS (m/z, %): 389 (M+, 30), 304 (77), 279 (20), 225 (20), 77 (100). Calcd. for C22H15NO4S (389.415): C, 67.86; H, 3.88; N, 3.60, S; 8.23. Found: C, 67.96; H, 3.95; N, 3.51, S; 8.30.
5-(Benzo[d]thiazol-2-yl)-3H-chromeno[2,3-b]azet- 3-ol (VIII). Method I. A solution of 3-formylchromone (III) (1 g, 0.003 mol), acetylacetone (0.328 mL, 0.003 mol), and ammonium acetate (0.25 g, 0.003 mol) in absolute ethanol (20 mL) was heated under reflux for 30 h. The precipitated solid after cooling was filtered off and crystallized from benzene to give chromenoazetol (VIII): red crystals, yield 52%, mp 228–230°C. IR spectrum: 3441 (br. OH), 3054 (CH aromatic), 2998 (CH aliphatic), 1606 (C=N). 1H NMR spectrum: 10.19 (s, 1H, OH, exchangeable with D2O), 8.07–7.91 (m, 4H, Ar-H), 7.52–7.35 (m, 3H, Ar-H), 6.95–6.92 (m, 2H, Ar-H + CH benzylic). 13C NMR spectrum: 167.94, 160.99, 154.18, 152.90, 139.51, 134.57, 129.51, 128.79, 126.90, 125.38, 124.51, 122.80, 122.76, 122.58, 116.56, 101.17, 90.18. MS (m/z, %): 306 (M+•, 5), 227 (100), 225 (23), 107 (54). Calcd. for C17H10N2O2S (306.343): C, 66.65; H, 3.29; N, 9.14, S; 10.48. Found: C, 66.78; H, 3.20; N, 9.22, S; 10.55.
5-(Benzo[d]thiazol-2-yl)-3H-chromeno[2,3-b]azet-3-ol (VIII). Method II. A solution of chromone derivative (VII) (0.6 g, 0.001mol) and ammonium acetate (0.118 g, 0.001 mol) in absolute ethanol (20 mL) was heated under reflux for 30 h. The precipitated solid after cooling was filtered off and crystallized from benzene to give (VIII). The compound identity was determined using identity mp, mixed mp, IR spectrum, and TLC comparison.
1-{2-(Benzo[d]thiazol-2-yl)-9,10a-dihydroxy-10a,H- benzo[c]chromen-8-yl}ethanone (IX). A solution of acetylacetone (0.328 mL, 0.003 mol) in acetic acid (5 mL) and concentrated hydrochloric acid (2–3 drops) was stirred at a temperature of 70–80°C for 15 min. A solution of 3-formylchromone (III) (1 g, 0.003 mol) in acetic acid was then added dropwise to the reaction mixture. The resulting solution was stirred for further 2 h at the same temperature. The dark red reaction mixture was cooled and then poured onto crushed ice. The solid deposit was filtered off, washed with water, dried, and recrystallized from benzene to give (IX): pale brown crystals, yield 43%, mp 205°C (decomp.). IR spectrum : 3450 (br. OH), 3064 (CH aromatic), 2949 (CH aliphatic), 1692 (C=O ketone). 1H NMR spectrum: 10.21 (s, 2H, 2 OH, exchangeable-D2O), 8.08–7.90 (m, 5H, Ar-H), 7.52–7.35 (m, 3H, Ar-H), 6.95–6.90 (m, 2H, Ar-H, C2-H chromone), 2.49 (s, 3H, CH3). MS (m/z, %): 389 (M+•, 2), 260 (18), 259 (100), 258 (25), 186 (14), 112 (17), 87 (19), 55 (53). Calcd. for 22H15NO4S (389. 415): C, 67.86; H, 3.88; N, 3.60, S; 8.23. Found: C, 67.74; H, 3.76; N, 3.69, S; 8.11.
Dihydrochromen-4-one derivatives (Xa) and (Xb). A solution of 3-formylchromone (III) (1 g, 0.003 mol) and p-aminoacetophenone or methyl p-aminobenzoate (0.003 or 0.006 mol) in absolute ethanol (20 mL) was heated under reflux for 30 min. The precipitated solid while hot in using p-aminoacetophenone or methyl p-aminobenzoate was filtered off and recrystallized from benzene to give (Xa) and (Xb) respectively.
2-(4-Acetylphenylamino)-3-((4-acetylphenylimino)-methyl)-6-(benzo[d]thiazol-2-yl)-2,3-dihydrochromen-4-one (Xa). Yellow crystals, yield 43 [1 : 1 mol] or 75% [1 : 2 mol], mp 136°C (decomp.). IR spectrum: 3280 (NH), 3059 (CH aromatic), 2956 (CH aliphatic), 1699 (C=O ketone), 1654 (C=O). 1H NMR spectrum: 11.98 (s, 1H, NH, exchangeable-D2O), 10.14 (s, 1H, CH=N), 8.90–6.56 (m, 15H, 15Ar-H), 6.29 (m, 1H, HC3-chromone), 2.98 (m, 1H, HC2-chromone), 2.37 (s, 6H, 2COCH3). MS (m/z, %): 559(M+•, 11), 304 (78), 279 (6), 235 (4), 227 (4), 120 (100). Calcd. for C33H25N3O4S (559.618): C, 70.83; H, 4.50; N, 7.51, S; 5.73. Found: C, 70.96; H, 4.63; N, 7.45, S; 5.68.
Methyl-4-(((6-(benzo[d]thiazol-2-yl)-2-((4-(methoxy-carbonyl)phenyl)amino)-4-oxochroman-3-yl)methylene)amino)benzoate (Xb). Yellow crystals, yield 45 [1 : 1 mol] and 78% [1 : 2 mol], mp 232–234°C. IR spectrum: 3299 (NH), 3059 (CH aromatic), 2946 (CH aliphatic), 1718 (C=Oester), 1654 (C=Ochromone), 1603 (C=N). 1H NMR spectrum: 11.97 (s, 1H, NH, exchangeable-D2O), 10.15 (s, 1H, CH=N), 8.72–6.97 (m, 17H, Ar-H), 5.93–5.91 (m, 1H, HC2-chromone), 3.88 and 3.84 (2s, 6H, COOCH3), 3.72 (m, 1H, HC3-chromone). MS (m/z, %): 591 (M+, 26), 402 (13), 273 (21), 188 (24), 227 (67), 43 (100). Calcd. for C33H25N3O6S (591.616): C, 66.99; H, 4.26; N, 7.10, S; 5.42. Found: C, 66.90; H, 4.14; N, 7.23; S, 5.55.
6-(Benzo[d]thiazol-2-yl)-3-((pyridin-4-ylimino)-methyl)-4H-chromen-4-one (XI). A solution of 3-formylchromone (III) (1 g, 0.003 mol) and 4-aminopyridine (0.349 g, 0.003 mol) in absolute ethanol was heated under reflux for 30 min. The precipitated solid while hot was filtered off and recrystallized from benzene to give (XI): yellow crystals, yield 72%, mp 177°C (decomp.). IR spectrum: 3058 (CH aromatic), 2952 (CH aliphatic), 1688 (C=Ochromone), 1645 (C=N). 1H NMR spectrum: 8.74–6.53 (m, 13H, 11Ar-H + HC2-chromone + CH=N). MS (m/z, %): 383 (M+, 1), 279 (9), 235 (2), 227 (100), 225 (2). Calcd. for C22H13N3O2S (383.415): C, 68.92; H, 3.41; N, 10.96, S; 8.36. Found: C, 69.05; H, 3.54; N, 10.88, S; 8.24.
1-{[6-(Benzo[d]thiazol-2-yl)-4-oxo-4H-chromen-3-yl]methylene}thiosemicarbazide (XII). A solution of 3-formylchromone (III) (1 g, 0.003 mol) and thiosemicarbazide (0.3 g, 0.003 mol) in absolute ethanol (20 mL) was heated under reflux for 30 min. The formed solid while hot was filtered off and recrystallized from ethanol to give thiosemicarbazide derivative (XII): yellow crystals, yield 69%, mp 222°C (decomp.). IR spectrum: 3212, 3133 (NH2 and NH), 3058 (CH aromatic), 2952 (CH aliphatic), 1646 (C=Ochromone), 1092 (C=S). 1H spectrum: 11.56 (s, 1H, NH, exchangeable with D2O), 9.22 and 8.71 (2s, 2H, CH=N, syn and anti isomers), 8.52 (d, 1H, benzothiazol-H, J = 2.4 Hz), 8.48 (d, 1H, benzothiazol-H, J = 2.4 Hz), 8.27 (s, 2H, NH2, exchangeable with D2O), 8.21–8.10 (m, 3H, Ar-H), 7.91 (d, 1H, benzothiazol-H, J = 9 Hz), 7.59 (d, 1H, benzothiazol-H, J = 9.9 Hz), 7.56–7.48 (m, 2H, HC2-chromone, syn and anti isomers). MS (m/z, %): 380 (M+•, 5), 304 (12), 279 (12), 253 (47), 225 (15), 197 (81). Calcd. for C18H12N4O2S2 (380.436): C, 56.83; H, 3.18; N, 14.73, S; 16.86. Found: C, 56.70; H, 3.29; N, 14.85, S; 16.90.
2-{2-[{6-(Benzo[d]thiazol-2-yl)-4-oxo-4H-chromen-3-yl}methylene]hydrazono}thiazolidin-4-one (XIII). A solution of thiosemicarbazide derivative (XII) (1 g, 0.002 mol) and chloroacetylchloride (0.383 g, 0.27 mL, 0.0023 mol) in glacial acetic acid (20 mL) in the presence of catalytic amount of fused sodium acetate was refluxed for 6 h. The reaction mixture was then cooled and poured onto ice/water. The formed solid was collected by filtration and recrystallized from benzene to give thiazolidinone derivative (XIII): yellow crystals, yield 55%, mp 210–211°C. IR spectrum: 3107 (NH), 3063 (CH aromatic), 2924 (CH aliphatic), 1701 (C=Othiazolidin), 1666 (C=Ochromone), 1621 (C=N). 1H NMR spectrum: 13.50 (s, 1H, OH, exchangeable with D2O), 11.11 (s, H, NH, exchangeable with D2O), 8.35 and 8.19 (2s, 2H, CH=N, syn and anti isomers), 8.13–7.91 and 7.54–7.17 (2 m, 9H, 7Ar-H+ 2 HC-2 chromone, syn and anti isomers), 6.92 (s, 1H, thiazolidinone-H). MS (m/z, %): 422 (M+2]+•, 4), 421 (M + 1]+•, 11), 420 (M+•, 31), 406 (35), 335 (68), 315 (16), 313 (100). Calcd. for C20H12N4O3S2 (420.457): C, 57.13; H, 2.87; N, 13.32, S; 15.25. Found: C, 57.01; H, 2.78; N, 13.41, S; 15.32.
6-(Benzo[d]thiazol-2-yl)chromeno[2,3-c]pyrazol-4-ol (XIV). Method I. A solution of 3-formylchromone (III), cyanoacrylate derivative (VI), dihydrochromen-4-one derivatives (Xa) and/or (Xb) (0.01 mol), and hydrazine hydrate (0.01 mol) in absolute ethanol (20 mL) was refluxed for 6–8 h and left to cool. The precipitated solid was then collected by filtration, dried, and recrystallized from benzene to afford chromeno[2,3-c]pyrazol (XIV) as yellow crystals, yield 56, 49, 50, and 51% using 3-formylchromone (III), cyanoacrylatederivative (VI), and dihydrochromen-4-one derivatives (Xa) and/or (Xb) respectively, mp 219–222°C. IR spectrum: 3406 (OH), 3054 (CH aromatic), 1605 (C=N). 1H NMR: 10.18 (s, 1H, OH, exchangeable with D2O), 8.11–6.91 (m, 8H, Ar-H).13C NMR spectrum: 167.95, 161.01, 154.21, 152.33, 142.76, 134.60, 129.51, 129.35, 128.77, 128.64, 126.86, 125.33, 124.55, 122.76, 122.51, 116.57, 80.51. MS (m/z, %): 319 (M+, 13), 304 (17), 279 (1), 253 (4), 227 (100), 197 (7). Calcd. for C17H9N3O2S (319.332): C, 63.94; H, 2.84; N, 13.16, S; 10.04. Found: C, 63.86; H, 2.77; N, 10.26; S, 10.12.
Method II. A solution of 3-formylchromone(III) (1 g, 0.003 mol) and thiosemicarbazide (0.29 g, 0.003 mol) in dimethylformamide (20 mL) was refluxed for 6 h. The reaction mixture was then poured onto ice-cooled water. The precipitated solid was collected by filtration, dried, and recrystallized from benzene to give chromeno[2,3-c]pyrazol (XIV): yield 51%.The compound identity was determined using identity mp, mixed mp, IR spectrum and TLC comparison.
Method III. A solution of thiosemicarbazide derivative (XII) (1 g, 0.002 mol) in DMF (20 mL) was refluxed for 6 h. The reaction mixture was then poured onto ice/cold water and the formed solid was collected by filtration, dried, and crystallized from benzene to give chromeno[2,3-c]pyrazol (XIV): yield 51%. The compound identity was determined using identity mp, mixed mp, IR spectrum and TLC comparison.
In Vitro Anticancer Activity Assays
Reagents and chemicals. Fetal bovine serum (FBS) and L-glutamine were obtained from Gibco Invitrogen Company (Scotland, UK). Dulbecco’s modified Eagle’s (DMEM) medium was provided by Cambrex (New Jersey, USA). Dimethyl sulfoxide (DMSO), doxorubicin, penicillin, streptomycin and sulfo-rhodamine-B stain (SRB) (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were obtained from Sigma Chemical Company (St. Louis, MO, USA). All other chemicals and reagents used in this study were of analytical grade and purchased from Sigma-Aldrich chemical Co. (St. Louis, MO, USA). The biological activity assay was performed at the Central Laboratory for Genetic Engineering, National Research Center, Dokki, Cairo, Egypt.
Cell lines. For anticancer activity screening of the tested compounds, lung A549, colon HCT116, breast MCF-7, prostate PC3, and liver HepG2 cancer cell lines, as well as the normal cell line (human normal melanocyte, HFB4), were obtained from the American Type Culture Collection (Rockville, MD, USA). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat inactivated fetal calf serum (GIBCO), penicillin (100 U/mL), and streptomycin (100 µg/mL) at 37°C in humidified atmosphere containing 5% CO2. Cells at a concentration of 0.50 × 106 were grown in a 25 cm2 flask in 5 mL of culture medium.
Cell viability assay. Anticancer activity of the tested compounds was measured in vitro using the sulfo-rhodamine-B stain (SRB) assay according to Skehanet al. [45]. Briefly, cells were inoculated in 96-well microtiter plate (104 cells/well) and incubated for 24 h before treatment with the tested compounds to allow attachment of cell to the wall of the plate. Test compounds were dissolved in DMSO at 1 mg/mL immediately before use and diluted to the appropriate volume just before addition to the cell culture. Different concentration of tested compounds and doxorubicin were added to the cells. Three wells were prepared for each individual dose. Monolayer cells were incubated with the compounds for 48 h at 37°C and in atmosphere of 5% CO2. After 48 h cells were fixed, washed, and stained for 30 min with 0.4% (w/v) SRB dissolved in 1% acetic acid. Unbound dye was removed by four washes with 1% acetic acid, and attached stain was recovered with Tris-EDTA buffer. Color intensity was measured in an ELISA reader. The relation between surviving fraction and drug concentration is plotted to get the survival curve for each cell line after the specified time. The concentration required for 50% inhibition of cell viability (IC50) was calculated and the results are given in Table 1. The results were compared to the effect of the reference drug doxorubicin.
CONCLUSION
The goal of the current research was to synthesize and assess antitumor activities of some novel 6-(benzothiazol-2-yl)-4-oxo-4H-chromene derivatives. The results indicated that compounds (Xa), (X), (XIII), and (XIV) exhibited hopeful in vitro antitumor activities against A594 (lung cancer) and HCT-116 (colon cancer), while only compound (XIV) displayed activity against MCF-7 (breast cancer) cells.
REFERENCES
Gottesman, M.M., Annu. Rev. Med., 2002, vol. 53, pp. 615–627.
Nobili, S., Landini, I., Giglioni, B., and Mini, E., Curr. Drug Targets, 2006, vol. 7, no. 7, pp. 861–879.
Li, J., Wang, X.L., Fang, Y.C., and Wang, C.Y., J. Asian Nat. Prod. Res., 2010, vol.12, no. 11, pp. 992–1000.
Patil, S. A., Wang, J., Li, X.S., Chen, J., Jones, T.S., Hosni-Ahmed, A., Patil, R., Seibel, W.L., Li, W., and Miller, D.D., Bioorg. Med. Chem. Lett., 2012, vol. 22, no. 13, pp. 44–58.
Kandeel, M.M., Kamal, A.M., Abdelall, E.K.A., and Elshemy, H.A.H., Der Pharma Chemica, 2012, vol. 4, no. 4, pp. 1653–1661.
Thomas, N. and Zachariah, S.M., Asian J. Pharm. Clin. Res., 2013, vol. 6, suppl. 2, pp. 11–15.
Zonouzi, A., Mirzazadeh, R., Safavi, M., Ardestani, S.K., Emami, S., and Foroumadi, A., Iranian J. Pharm. Res. (IJPR),2013, vol. 12, no. 4, pp. 679–685.
Reddy, K.R., Rao, P.S., Dev, G.J., Poornachandra, Y., Kumar, C.G., Rao, P.S., and Narsaiah, B., Bioorg. Med. Chem. Lett., 2014, vol. 24, no. 7, pp. 1661–1663.
Rahmani-Nezhad, S., Safavi, M., Pordeli, M., Ardestani, S.K., Khosravani, L., Pourshojaei, Y., Mahdavi, M., Emami, S., Foroumadi, A., and Shafiee, A., Eur. J. Med. Chem., 2014, vol. 86, pp. 562–569.
Fouad, S.A., Int. J. Adv. Res., 2014, vol. 2, no. 12, pp. 442–453.
Banerjee, S., Wang, J., Pfeffer, S., Ma, D., Pfeffer, L.M., Patil, S.A., Li, W., and Miller, D.D., Molecules, 2015, vol. 20, pp. 17152–17165.
Puppala, M., Zhao, X., Casemore, D., Zhou, B., Aridoss, G., Narayanapillai, S., and Xing, C., Bioorg. Med. Chem., 2016, vol. 24, no. 6, pp. 1292–1297.
Fouda, A.M., Med. Chem. Res., 2016, vol. 25, pp. 1229–1238.
Salem, M.S., Marzouk, M.I., Ali, S.N., and Madkour, H.M. F., Eur. J. Chem., 2012, vol. 3, no. 2, pp. 220–227.
Yadav, P.S., Devprakash, and Senthilkumar, G. P., Int. J. Pharm. Sci. Drug Res., 2011, vol. 3, no. 1, pp. 1–7.
Abbas, E.M.H., Salem, M.S., Kassem, A.F.M., Abd El-Moez, Sh.I., and El-Kady, M., Eur. J. Chem., 2015, vol. 6, no. 2, pp. 98–106.
Kok, S.H., Gambari, R., Chui, C.H., Yuen, M.C.W., Lin, E., Wong, R.S.M., Lau, F.Y., Cheng, G.Y.M., Lam, W.S., Chan, S.H., Lam, K.H., Cheng, C.H., Lai, P.B.S., Yu, M.W.Y., Cheung, F., Tang, J.C.O.,and Chan, A.S.C., Bioorg. Med. Chem., 2008, vol. 16, no. 7, pp. 3626–3631.
Havrylyuk, D., Mosula, L., Zimenkovsky, B., Vasylenko, O., Gzella, A., and Lesyk, R., Eur. J. Med. Chem., 2010, vol. 45, no. 11, pp. 5012–5021.
Devmurari, V.P., Shivanand, P., Goyani, M.B., Nandanwar, R.R., Jivani, N.P., and Perumal, P. Int. J. Chem. Tech. Res., 2010, vol. 2, no. 1, pp. 681–689.
Furlan, A., Colombo, F., Kover, A., Issaly, N., Tintori, C., Angeli, L., Leroux, V., Letard, S., Amat, M., Asses, Y., Maigret, B., Dubreuil, P., Botta, M., Dono, R., Bosch, J., Piccolo, O., Passarella, D., and Maina, F., Eur. J. Med. Chem., 2012, vol. 47, pp. 239–254.
Shi, X., Wang, Z., Xia, Y., Ye, T., Deng, M., Xu, Y., Wei, Y., and Yu, L., Molecules, 2012, vol. 17, pp. 3933–3944.
Caputo, R., Calabrò, M.L., Micale, N., Schimmer, A.D., Ali, M., Zappalà, M., and Grasso, S., Med. Chem. Res., 2012, vol. 21, no. 9, pp. 2644–2651.
Lindgren, E.B., de Brito, M.A., Vasconcelos, T.R.A., de Moraes, M.O., Montenegro, R.C., Yoneda, J.D., and Leal, K.Z., Eur. J. Med. Chem., 2014, vol. 86, pp. 12–16.
Gurdal, E.E., Buclulgan, E., Durmaz, V., Cetin-Atalay, R., and Yarim, M., Anticancer Agents Med. Chem., 2015, vol. 15, no. 3, pp. 382–389.
El-Damasy, A.K., Lee, J.H., Seo, S.H., Cho, N.C., Pae, A.N., and Keum, G., Eur. J. Med. Chem., 2016, vol. 115, pp. 201–216.
Singh, M., Singh, S.K., Thakur, B., Ray, P., and Singh, S.K., Anticancer Agents Med. Chem., 2016, vol. 16, no. 6, pp. 722–739.
Meenakshi, S., Arusha, M., Gopeshwar, N., and Sushil, S.K., Anti-Cancer Drugs, 2016, vol. 27, no. 6, pp. 519–532.
Mohamed, K.S., Refat, H.M., and Mohamed, N.A.H., Heterocycles, 2016, vol. 92, no. 8, pp. 1415–1429.
Bozdag-Dundar, O., Evranos, B., Das-Evcimen, N., Sarhkaya, M., and Ertan, R., Eur. J. Med. Chem., 2008, vol. 43, no. 11, pp. 2412–2417.
Havrylyuk, D., Zimenkovsky, B., Vasylenko, O., Day, C.W., Smee, D.F., Grellier, P., and Lesyk, R., Eur. J. Med. Chem., 2013, vol. 66, pp. 228–237.
Kapoor, G., Pathak, D.P., Bhutani, R., and Kant, R., J. Chem. Pharm. Res., 2016, vol. 8, no. 4, pp. 151–168.
Salem, M.S., and Ali, M.A.M., Biol. Pharm. Bull., 2016, vol. 39, pp. 473–483.
El-Hashash, M.A.M., Salem, M.S., and Al-Mabrook, S.A.M., Res. Chem. Intermed., 2018, vol. 44, pp. 2545–2559.
Pal, D., Saha, S., and Singh, S., Int. J. Pharm. Pharm. Sci., 2012, vol. 4, no. 2, pp. 98–104.
Jamwal, A., Javed, A., and Bhardwaj, V., J. Pharm. Bio. Sci., 2014, vol. 3, pp. 114–123.
Ali, A.R., El-Bendary, E.R., Ghaly, M.A., and Shehata, I.A., Eur. J. Med. Chem., 2014, vol. 75, pp. 492–500.
Lacova, M., Loos, D., Furdik, M., Matulova, M., and El-Shaaer, H.M., Molecules, 1998, vol. 3, pp. 149–158.
Hu, K., Yang, Z., Pan, S., Xu, H., and Ren, J., Eur. J. Med. Chem., 2010, vol. 45, pp. 3453–3458.
Altıntop, M.D., Temel, H.E., Sever, B., Çiftçi, G.A., and Kaplancıkl, Z.A., Molecules, 2016, vol. 2, pp. 1598–1617.
Hassan, G., El-Messry, S., Al-Omary, F., and El-Sabbagh, H., Bioorg. Med. Chem. Lett., 2012, vol. 22, no. 20, pp. 6318–6323.
Amit, C., Sheelmani, S.A., Payal, C., and Dhawan, R.K. Adv. J. Pharm. Life Sci. Res., 2014, vol. 2, no. 1, pp. 1–7.
Spanò, V., Attanzio, A., Cascioferro, S., Carbone, A., Montalbano, A., Barraja, P., Tesoriere, L., Cirrincione, G., Diana, P., and Parrino, B., Mar. Drugs, 2016, vol. 14, pp. 226–244.
Das, D., Banerjee, R., and Mitra, A., J. Chem. Pharm. Res., 2014, vol. 6, no. 11, pp. 108–116.
Silva, M.S., Pinto, D.C.G.A., Cavaleiro, J.A.S., Levai, A., and Patonay, T., Arkivoc, 2004, vol. VII, pp. 106–123.
Skehan, P., Storeng, R., Scudiero, D., Monks, A., McMahon, J., Vistica, D., Warren, J.T., Bokesch, H., Kenney, S., and Boyd, M.R., J. Nati. Cancer Inst., 1990, vol. 82, pp. 1107–1112.
ACKNOWLEDGMENTS
Authors would like to express their deep appreciation and indebtedness to Chemistry Department, Faculty of Science, Ain Shams University, for their support of the work.
Author information
Authors and Affiliations
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
The article is published in the original.
Corresponding author: e-mail: marwa_k@sci.asu.edu.eg.
Rights and permissions
About this article

Cite this article
Eman A. El-Helw, Derbala, H.A., El-Shahawi, M.M. et al. Synthesis and In Vitro Antitumor Activity of Novel Chromenones Bearing Benzothiazole Moiety. Russ J Bioorg Chem 45, 42–53 (2019). https://doi.org/10.1134/S1068162019010047
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162019010047