
Abstract
The results of the study of the shifts of the X-ray Kα and Kβ lines of manganese and copper obtained for the nanoparticles of the oxides CuO, MnO, Mn3O4, and MnO2 (relative to the corresponding bulk materials) are presented. The oxide nanoparticles are synthesized in the pores of borosilicate glass with an average pore diameter of 7 ± 1 nm from the corresponding nitrates introduced into the pores by capillary impregnation. It is found that a small (of about ~0.1 electron/atom) increase in the role of 3d electrons is observed for the nanocomposites with CuO and Mn3O4, while a small decrease in the participation of these electrons in the chemical bond is observed for the nanocomposites with MnO2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 INTRODUCTION
The investigation of the properties and structure of nanoparticles has been attracting close attention in recent years in view of the development of fundamentally new technologies utilizing ultradispersed materials and composites based on them. By now, it has been experimentally found that the macroscopic physical properties and crystal structure of a whole series of such objects significantly differ from the properties of similar bulk materials. There are many methods for obtaining nanoparticles, and the introduction (or chemical synthesis) of substances into the pores of natural or artificial matrices is one of them. Thus, e.g., new, previously unknown crystal phases have been found for gallium [1, 2] and indium [3, 4] nanoparticles in nanoporous glass. Nanocomposite materials (NCMs) obtained based on porous matrices possess a whole range of macroscopic properties attractive for practical application such as the high dielectric permeability [5, 6], growth in the ionic conductivity [7, 8], expansion of the range of existence of the ferroelectric phase [9, 10], and sharp increase in the critical magnetic fields for the superconducting state [11, 12]. It has been found that substantial transformation of the elementary excitation spectra and a change in the constants of coercive bonding between atoms occurs in ultrasmall metal particles [13–16]. Naturally, the properties of the electron subsystems of ultradispersed substances were not overlooked as well; thus, it was shown that, in metal nanoparticles with an average size of 1–2 nm and in structures of the “core–shell” type [17–20], a size-induced metal–dielectric transition occurs, the density of states near the Fermi level changes, the contribution from the electron subsystem to the thermal capacity [21] at low temperatures increases, shifts of the 3d and 4d electron levels occur [20, 22, 23], etc. It is natural to expect that the reorganization of the electron subsystem will also lead to a change (a shift) in the energy of the X-ray radiation lines characteristic for this material. The procedure for measuring small energy shifts of X-ray lines by crystal diffraction spectrometry [24, 25] lies in the fact that this shift of the levels is measured directly and under the same experimental conditions rather than found as a difference of two large numbers that correspond to the measured energies of the X-ray lines for two samples under comparison. The sensitivity of the method makes it possible to directly measure the values of the shifts of the lines of about 10–3–10–4 from their natural width (i.e., a value of ~1 meV). Earlier, the shifts of the X-ray K lines in heavy elements (z = 32–74) upon the transition from one chemical compound to another (with different valences) which had been considered experimentally unmeasurable until then were found by this method for the first time [24]. Moreover, the method gives the possibility to unambiguously identify the type of the electrons participating in the chemical bond and to determine the occupancies of the outer (valence) orbitals of the atoms with a high accuracy, i.e., to microscopically study the electron structures of atoms because the change in the occupancy of the valence states in particular leads to a small change in the energies of the internal X-ray transitions of this atom [26–28]. Assuming that such small shifts may be present for nanoparticles and/or nanostructures based on nanoporous matrices, we decided to apply this method to the investigation of NCMs as well. The first results obtained for the nanoparticles of In, Pb, Pd, and some manganese oxides [4, 29, 30] showed that the shifts being observed are quite observable and statistically confident despite their smallness. The aim of this work was the execution of more detailed studies of the effect of confined geometry (or size effect) on the shifts of the X-ray K lines of manganese and copper in the oxides of these elements in comparison with similar results for bulk materials.
2 SAMPLES AND MEASUREMENT PROCEDURE
Porous sodium borosilicate glass (PG7) with an average pore diameter of 7(1) nm fabricated at the Ioffe Institute was used as the initial matrices. In this glass, the pores form a multiconnected dendritic through network of channels; the total porosity of the used glass was 22–25%. The average pore diameter was determined by mercury porosimetry. Thin plates with a thickness of about 1 mm were fabricated from the porous glass which were used for further filling and preparation of nanoparticles.
The introduction of manganese oxides (MnO, MnO2, Mn3O4) and copper oxide (CuO) into the pores was executed in several stages. The plates of PG7 porous glass were impregnated with saturated aqueous solutions of Mn(NO3)2 and Cu(NO3)2, respectively, under the action of capillary forces. Then the samples were dried and calcinated in air at 250°C for 30 min; here, nitrates in the pores of PG7 were decomposed to insoluble oxides (as well as oxo and hydroxo nitrates) with no defined chemical composition which further acted as the precursors for the synthesis of the target substances. The procedures of capillary impregnation and calcination were periodically repeated to achieve the required degree of filling of the pores.
To synthesize the target compounds in the pores of PG7, the samples were calcinated in various gaseous atmospheres. Thus, the oxides CuO and MnO2 were synthesized by calcinating the samples filled with the corresponding precursors in oxygen, MnO, in hydrogen, and Mn3O4, in forevacuum (at a partial pressure of oxygen of ~10–2 Torr). All the calcinations were performed at 400°C for 5 h. The filling of the pores of PG7 with the oxides was about 30–40% of the total pore space.
Prior to the measurement, the plates were ground to a powder that was placed into special capsules for the studies. The measurements of the shift of the Kα1, Kα2, and Kβ1,3 lines for bulk and nanostructured manganese oxides were performed at room temperature on a crystal diffraction spectrometer of the Johann type [25] under the same experimental conditions. The relative shift of the lines between the bulk and nanostructured samples was determined in the experiments. The measuring and processing procedure is described in detail in [25]. The obtained values of the shifts of the X-ray K lines are presented in Table 1. All the shifts were determined relative to a reference samples, i.e., e.g., the value of the shift for a Mn–Mn2O3 pair ΔE(Kα1) = EKα1(Mn) – EKα1(Mn2O3), this shift is denoted as shift(ΔE(Kα1)) in Table 1; the headings of the columns in this same table, shift(ΔE(Kα2)) and shift(ΔE(Kβ1,3)), correspond to the shifts of the Kα2 and Kβ1,3 lines. The oxide Mn2O3 served as the reference sample (hereinafter, Sref) for almost all the bulk manganese oxides. For the nanostructured Mn oxides, the corresponding Sref was a similar bulk oxide. A sample of bulk metal was used as Sref for copper oxides. Table 1 presents the results averaged by several series of measurements (generally about ten series).
All the obtained samples were preliminarily tested on a DRON-2 X-ray diffractometer on a Kα radiation of copper.
3 RESULTS AND DISCUSSION
3.1 Diffraction Spectra for the NCMs Based on PG7
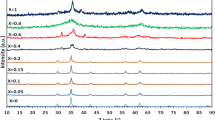
Figures 1a–1c present the diffraction patterns for the bulk (the bottom curves in each figure) and synthesized in the pores of PG7 (the top curves) MnO, Mn3O4, and MnO2.

(a, b) Experimental diffraction spectra of bulk and nanostructured manganese oxides and (c) spectra for α-MnO2 and ε-MnO2.
The experimental results are highlighted in gray, and the reflections corresponding to the structures of manganese oxides obtained as a result of fitting are outlined by the black lines for the illustrative purposes. It is well seen that the structures of MnO and Mn3O4 in the pores of PG7 (Figs. 1a, 1b) well correspond to the structures of the bulk oxides. The additional peak in the region of 2θ ~ 20° is associated with the scattering on amorphous SiO2 which constitutes a PG7 matrix itself.
As it was shown by the results of the profile analysis, a PG7 + MnO2 sample (Fig. 1c) is a mixture of “common” α-MnO2, the same as in the bulk sample, and, to an even greater extent, a relatively rarely occurring ε-MnO2—the modeled spectra for these two phases are presented in Fig. 1c at the bottom. Similar results were obtained (Fig. 2) for bulk copper oxide (the bottom curve) and a PG7 + CuO NCM (the top curve).

Experimental diffraction spectra for bulk and nanostructured copper oxides. The background is subtracted in the case of bulk copper oxide.
It should be noted that the diffraction peaks are significantly broadened for all the NCMs in comparison with the bulk materials; we obtained the estimate of the sizes of the oxide nanoparticles in the pores from the analysis of this broadening which turned out to be of 10–12 (for manganese oxides) to 14.5 nm for CuO in the pores of PG7.
These values are in good agreement with the sizes obtained from the analysis of the data on the diffraction of neutrons on these same samples [31, 32]. No other extraneous peaks were observed in the diffraction spectra, which gives the evidence of the fact that the particular compounds of interest to us are present in the fabricated NCMs.
3.2 X-Ray Spectroscopy of the Chemical Shifts of the K Lines of the Oxides
The method for shifting X-ray lines (differential X‑ray emission spectroscopy) developed in [33] is universal and possesses high selectivity, which makes it possible to unambiguously and independently identify the type of the electron (s, p, d, f ) participating in the chemical bond; this method also makes it possible to determine the occupancies of valence orbitals with a high accuracy (up to 0.01 electron/atom). Another advantage of this approach is the fact that the obtained results refer to the entire volume of the samples and not only to its surface. When a valence electron is spent for the formation of a chemical bond, a small but, as it has been shown in [26, 33], quite measurable energy shift of the Kα1,2 (1s–2p), Kβ1,3 (1s–3p), and Kβ2,4 (1s–4p) lines occurs. It was shown in this same review that the electron state of an element in a compound can be characterized by an original “facsimile”—a set of the shifts of the diagram X-ray lines relative to the corresponding metal. In a general case, the value of the shift (facsimile) depends on the valence, degree of ionicity (covalency) of the element in the compound, and spin state of the d shell. The closeness of the facsimile of any element in two its compounds evidences the identity of the electron configurations (valence, ionicity, and covalency) of this (under study) element in these compounds.
A pertinent question arises what shifts of the X-ray lines can be expected in the case of ultradispersed nanoparticles and/or compounds which are under the conditions of confined geometry. First of all, it should be noted that a large fraction of surface atoms (up to 30%) is characteristic for such samples (let us assume so far that the matrix does not participate in the modification of the properties of the introduced materials but only forms the conditions of confined geometry) depending on the shape of the nanoparticles. Second, it has been found [13, 14, 34] that the local symmetry in the surface layers of a nanoparticle (up to the third coordination sphere inclusive) can strongly differ from the symmetry characteristic for the bulk material, which leads to, e.g., significant modification of the phonon spectra [13–16], and, here, the dependence on the size of the nanoparticles is clearly seen [13, 14]. Naturally, this may (and will) lead to the distortion of the wave functions of the outer electrons of the atoms in the near-surface layers of the nanoparticles. Moreover, in the case when such distortions of the elementary cells are relatively large and spread to a noticeable depth, a change in the symmetry of the crystal lattice is possible which may lead to a change in the mutual positions of the levels of the valence orbitals and zones, which, in turn, will cause the redistribution of the electrons between the zones in the metals and change in the covalency in the insulators. In these cases, noticeable effects of the shifts of the internal levels and X-ray lines are possible. In the case of significant distortion of the symmetry of the crystal lattice (i.e., a change in the internal crystal field as well), a change in the spin state of the atoms is also possible which can be observed by the change in the split of the Kβ1,3 and Kβ' lines Δ(E(Kβ1,3) –E(Kβ')) which is denoted as Split(Kβ1,3 – Kβ') in Table 1 for the studied pairs of samples presented in column 2.
3.2.1. Shifts of the K lines in copper oxides. According to the electron structure, there is one 4s electron in addition to the filled 3d shell in metallic copper, because of which the values of the shifts of the K lines of copper (relative to the metal) provide the 4s facsimile for a monovalent compound with high ionicity, and 3d facsimile, for a bivalent compound. These two parameters were earlier determined in [28]. Figure 3a presents the measured ΔE(Kα1), ΔE(Kα2), and ΔE(Kβ1,3) shifts for bulk CuO and Cu2O and a CuO + PG7 NCM, and the inset at the bottom, the differences ΔE(CuOnano – CuObulk) for the Kα1, Kα2, and Kβ1,3 lines. Figure 3b presents the facsimile for the 4s and 3d electrons of copper and, for comparison, nickel [28].

(a) Experimental shifts of the K lines for copper oxides and (b) facsimile of the 4s and 3d electrons of copper adopted from [28].
It is seen from the figure that the chemical bond in Cu2O is executed by 4s electrons, and the ionicity is not high here because the shifts for Cu2O are significantly smaller in magnitude when compared to the 4s facsimile. In the process of further oxidation (formation of CuO), the remaining 4s electrons are additionally gathered, and 3d electrons become involved in the bond in accordance with the ionicity. The total effect is relatively small due to the opposite signs of the facsimiles (shifts) of the 4s and 3d electrons (Fig. 3b). Indeed, if one 4s electron and one 3d electron were fully spent for the formation of the chemical bond in CuO, the shift being observed (according to the facsimiles of these electrons) should have been, e.g., for Kα1, ΔE(Kα1) = 189 – 110 = 79 (5) meV. In the experiment, we observe a shift of 105.5 ± 3.1 meV, i.e., no full involvement of the 3d electron into the chemical bond occurs.
As is seen from Fig. 3a, the shifts of the K lines of copper in a CuO + PG7 NCM are somewhat lower than in bulk CuO, i.e., the negative contribution becomes greater due to the 3d electron. Here, the difference of the shifts ΔE(CuOnano – CuObulk) (the facsimile of a “pure” ultradispersion effect (Fig. 3a, inset)) is very similar to the 3d facsimile in shape. The change (the degree of involvement of the 3d electron) can be estimated as ΔE(CuOnano – CuObulk)/ΔE (the 3d facsimile of Cu) using the data for Kα1 and Kα2, which gives the value of Δn(3d) = 0.13 ± 0.03 el/atom. This means that by 0.13 ± 0.03 3d electron/atom more participate in the chemical bond in a CuO + PG7 NCM when compared to bulk CuO. It should be noted that the facsimiles used henceforward have been experimentally obtained, and the ionicities and/or covalencies of the compounds being used have not been taken into account during the calculation, while these factors may change the values of the presented “facsimile” shifts, i.e., the estimates of Δn(3d) as well, apparently, up to 20–30%. Let us emphasize that the greater in magnitude shifts of the K lines of copper for the NCM in comparison with the bulk material by no means mean higher valence. The valence and oxidation state of Cu in CuO was and remains to be two, just the ionicity (in other words, degree of covalency and hybridization) of this chemical bond changes.
3.2.2. The shift of the K lines for manganese oxides. Figures 4a–4c present the experimental shifts of the X-ray K lines of manganese depending on the type of the lines, valence of Mn, and split of Kβ1,3–Kβ' lines for the oxides MnO, Mn3O4, and MnO2.

Experimental dependences of the shifts of the X-ray K lines of manganese on (a) type of the lines and (b) valence as well as (c) split of the Kβ–Kβ' lines for bulk and nanostructured oxides MnO, Mn3O4, and MnO2.
It is seen from Fig. 4a that the shifts being observed for manganese oxides are substantially different in shape when compared to copper oxides (Fig. 3a) but similarly depend on the valence—an increase in the valence leads to the negative shifts of all the lines. Here, both the shifts of the K lines and split of the Kβ1,3–Kβ' lines almost linearly depend on the value of the valence of manganese in these compounds, which is determined by the relationship of these parameters with the occupancy of the 3d orbitals. Note that the high values of the split of the Kβ1,3–Kβ' lines give the evidence of the high-spin state of the atoms (ions) of manganese in these oxides which is confirmed by the published data on the magnetic moments of manganese and splits of the Kβ1,3–Kβ' lines and 3s level of similar atoms [35–39].
As it can be seen from Fig. 4c, ultradispersion of manganese oxides does not lead to any change in the spin (i.e., magnetic as well) state of the Mn atoms because the values of the split of the Kβ1,3–Kβ' lines of NCMs and similar bulk samples are in fact within one measurement error. Note that, in fact, the effect of the change in the spin state is very great, thus, e.g., the change in the high-spin state of bivalent manganese in MnO (the total spin S = 5/2) to the medium-spin state (S = 3/2) would have led to a decrease in Split(Kβ1,3–Kβ') from ~14.5 to ~13 eV (Fig. 4c). Indeed, it would have been impossible not to notice such changes. The commentary concerning the spin state is presented in part 3.2.3 of this work.
The manifestation of ultradispersion is clearly seen in Fig. 4a—the shifts for the NCMs and bulk samples noticeably differ in almost all the points. To obtain a pure effect, we performed direct measurements of the shifts ΔE (nano–bulk), the results of which are presented in Fig. 5a. As it can be seen from Fig. 5a, statistically significant changes in the shifts of the X-ray K lines of Mn, with different directionalities in a general case, are observed for manganese oxides upon the transition from bulk to nanostructured samples. To clarify the physicochemical sense of such changes, they should have been compared to the facsimile of the 4s and 3d electrons of manganese but there are none yet, and those available for Cu and Ni (Fig. 3b) would hardly be suitable due to the range of the distance from Mn to Cu and actually oppositeness of the spin (magnetic) states.

Change in the shifts of the K lines of manganese in the case of the (a) ultradispersion of its oxides and (b) change in the valence of manganese.
However, as it was indicated above, all the changes in the valence state in the oxides under consideration are determined by the occupancy of the 3d orbitals only. Because of this, it can be assumed that the changes in the chemical bond upon ultradispersion will also be determined by the degree of participation of the 3d electrons in particular in it. Therefore, just the facsimile of the 3d electrons of manganese will be sufficient for us which can apparently be obtained from the difference of the shifts of the K lines of manganese of two oxides with different valences. Such differences ΔE(Mn3O4 – MnO) and ΔE(MnO2 – Mn3O4) are presented in Fig. 5b, from which it can be seen that they are similar in shape with good accuracy, and the ratio of the shifts ΔE(MnO2 – Mn3O4) to ΔE(Mn3O4 – MnO) is r(ΔE) = 1.71 ± 0.07. Since the ratio of the differences of the valences r(Δ\({v}\)) = Δ\({v}\)(MnO2 – Mn3O4)/Δ\({v}\)(Mn3O4 – MnO) = 1.33/0.67 = 2, apparently, it can be stated that the difference of r(ΔE) from two is determined by the decrease in the ionicity of the manganese atoms with the increase in its valence in the oxides. This gives the ground to set, in the first approximation, the curves in Fig. 5b as the facsimile of the 3d electrons of manganese with respect to the shape but underestimated with respect to the value. It can apparently be concluded from the comparison of the curves in Figs. 5a and 5b that the facsimile for the NCMs with Mn3O4 and facsimile of the 3d electrons of manganese have close (Λ‑like) shapes, from where it can be concluded that a large fraction of 3d electrons participates in the crystallochemical bond in a Mn3O4 + PG7 NCM in comparison with the bulk material. From the ratios of the shifts of these curves, the estimate of this fraction is Δ3d(Mn3O4) = 0.21 ± 0.06 electron/atom. Let us remind that such facsimiles of the 3d electrons of manganese have an underestimated value, i.e., the obtained value of Δ3d(Mn3O4) is most likely overestimated. The same also refers to other cases. In the cases of MnO and MnO2, the facsimiles of the difference between the NCMs and bulk samples are reciprocal in shape to the facsimiles of the 3d electrons (V-shaped), which corresponds to a decrease in the participation of the 3d electrons of manganese upon the ultradispersion of these oxides. The estimates of the values of these changes are Δ3d(MnO) = –0.05 ± 0.09 electron/atom and Δ3d(MnO2) = –0.11 ± 0.01 electron/atom, and the large error of the estimate in the first case is to a significant extent associated with the noticeable difference in the shape of the curves for MnO in Fig. 5a and facsimiles of the 3d-electrons in Fig. 5b.
3.2.3. The spin state. In a general case, the 3d atoms in compounds have a magnetic moment. The magnetic states of an atom are divided to high-spin (e.g., the 3d(↑)5 electron configuration, magnetic moment μ ≈ 5 μB for MnO), medium-spin (3d(↑)43d(↓)1, μ ≈ 3 μB), and low-spin (3d(↑)33d(↓)2, μ ≈ 1 μB) with respect to its value. The high- and low-spin states of the 3d atoms are even visibly distinguishable by the shape of the spectrum in the region of the (Kβ1,3–Kβ') lines (Fig. 6); their quantitative evaluation (Split(Kβ1,3–Kβ')) can also be obtained from the suitable approximation of this spectrum. It is most often considered that this split is determined by the exchange interaction between the 3p and 3d shells. Because of this, the value of the split is exclusively associated with the total spin, i.e., magnetic moment, of the atom. The macroscopic (cooperative) state of matter (ferromagnetic, antiferromagnetic, paramagnetic, etc.) has almost no effect on Split(Kβ1,3–Kβ').

Profile of the Kβ line of a 3d element indicating the parameters of Split(Kβ1,3–Kβ') and Shift(Kβ1,3) presented in the text and Table 1. L-spin is the low-spin state and H‑spin is the high-spin state.
4 CONCLUSIONS
It has been shown that the crystal structure of the oxides MnO, Mn3O4, and CuO in the bulk state and NCMs based on PG7 is almost the same; a mixture of phases, α-MnO2 generally intrinsic to the bulk material and quite exotic ε-MnO2, is observed in the case of a PG7–MnO2 NCM. It has been found from the analysis of the shifts of the X-ray K lines that a small (of ~0.1 electron/atom) increase in the role of the 3d electrons in the chemical bond is observed in the NCMs with CuO and Mn3O4, while a small decrease in the participation of the 3d electrons in the chemical bond is observed in a PG7 + MnO2 NCM. The situation with the NCMs containing MnO nanoparticles is still ambiguous and requires further investigation and analysis because it is impossible to rule out a significant change in the covalency in comparison with the bulk sample. The common trend of the values of the shifts of the K lines for bulk oxides corresponds to those expected in the case of a change in the valence of manganese from 0 to +4 based on the approach presented in [24]. Here, the values of the shifts of all the K lines linearly depend on the valence of manganese, and the negative value of the inclination of these straight lines clearly evidences that the increase in the oxidation state of Mn is determined by the decrease in the occupancy of the 3d orbital. The differently directed shift of the K lines of manganese upon the transition from the metal to the monoxide is well explained by the fact that, in this case, the chemical bond is executed by a mixture of 3d and 4s states. It is also well seen from Table 1 that a significant statistically confident difference in the energy shifts of the K lines is present between nanostructured and massive manganese oxides with the same valence. It should be emphasized that these effects have no regular dependence on what electron is spent for the formation of the bond. Apparently, the occurring reorganization of the electron system is associated not only with the ultradispersion (with the size of the nanoparticles) but also with the characteristic features of the interaction of the nanoparticles of manganese oxides (possibly, copper as well) with the matrix itself. Here, it should be noted that the used method gives the results averaged throughout the volume of the nanoparticles. At the same time, it is natural to assume that the electron properties of the internal areas of the nanoparticles do not differ from the properties of the bulk materials, and all the differences are observed in the interface layer. The thickness of this surface layer with the environment changed in comparison with the bulk material can be set equal to the radius of the third coordination sphere according to [13, 14, 34]; in such a case, in our NCMs (taking into account their diffraction size), up to 30% atoms can be present in these layers. Because of this, the obtained values should be considered the lower-bound estimates.
REFERENCES
I. G. Sorina, C. Tien, E. M. Charnaya, Yu. A. Kumzerov, and L. A. Smirnov, Phys. Solid State 40, 1407 (1998).
E. V. Charnaya, C. Tien, K. J. Lin, and Yu. A. Kumzerov, Phys. Rev. B 58, 11809 (1998).
B. Balamurugan, F. E. Kruis, S. M. Shivaprasad, O. Dmitrieva, and H. Záhres, Appl. Phys. Lett. 86, 083102 (2005).
A. A. Naberezhnov, A. E. Sovestnov, and A. V. Fokin, Tech. Phys. 56, 637 (2011).
S. V. Pan’kova, V. V. Poborchii, and V. G. Solov’ev, J. Phys.: Condens. Matter 8, L203 (1996).
E. V. Colla, E. Yu. Koroleva, Yu. A. Kumzerov, B. N. Savenko, and S. B. Vakhrushev, Ferroelectr. Lett. 20, 143 (1996).
J. Maier, Nat. Mater. 4, 805 (2005).
N. Sata, K. Eberman, K. Eberl, and J. Maier, Nature (London, U.K.) 408, 946 (2000).
S. V. Baryshnikov, E. V. Charnaya, A. Yu. Milinskiy, Yu. A. Shatskaya, C. Tien, and D. Michel, Phys. B (Amsterdam, Neth.) 405, 3299 (2010).
A. Naberezhnov, E. Koroleva, A. Sysoeva, S. Vakhrushev, E. Rysiakiewicz-Pasek, and M. Tovar, in Proceedings of 6th International Conference on Broadband Dielectric Spectroscopy and Applications, Madrid,2010, p. 126.
G. Kh. Panova, A. A. Naberezhnov, and A. V. Fokin, Phys. Solid State 50, 1370 (2008).
A. A. Shikov, M. G. Zemlyanov, P. P. Parshin, A. A. Naberezhnov, and Yu. A. Kumzerov, Phys. Solid State 54, 2345 (2012).
P. M. Derlet, R. Meyer, L. J. Lewis, U. Stuhr, and H. van Swygenhoven, Phys. Rev. Lett. 87, 205501 (2001).
P. M. Derlet and H. van Swygenhoven, Phys. Rev. Lett. 92, 035505 (2004).
P. P. Parshin, M. G. Zemlyanov, G. Kh. Panova, A. A. Shikov, A. A. Naberezhnov, Yu. A. Kumzerov, I. V. Golosovsky, and A. S. Ivanov, J. Exp. Theor. Phys. 111, 996 (2010).
P. P. Parshin, M. G. Zemlyanov, G. Kh. Panova, A. A. Shikov, Yu. A. Kumzerov, A. A. Naberezhnov, I. Sergeev, W. Crichton, A. I. Chumakov, and R. Ruffer, J. Exp. Theor. Phys. 114, 440 (2012).
M. Rosenblit and J. Jortner, J. Phys. Chem. 98, 9365 (1994).
R. Busani, M. Folkers, and O. Chesnovsky, Phys. Rev. Lett. 81, 3836 (1998).
C. P. Vinod, G. U. Kulkarni, and C. N. R. Rao, Chem. Phys. Lett. 289, 329 (1998).
C. N. R. Rao, G. U. Kulkarni, A. Govindaraj, B. C. Satishkumar, and P. G. Thomas, Pure Appl. Chem. 72, 21 (2000).
A. A. Shikov, G. Kh. Panova, M. G. Zemlyanov, P. P. Parshin, Yu. A. Kumzerov, A. A. Naberezhnov, and D. S. Shaitura, Phys. Solid State 53, 2515 (2011).
J. N. Andersen, D. Hennig, E. Lundgren, M. Methfessel, R. Nyholm, and M. Scheffle, Phys. Rev. B 50, 17525 (1994).
Chang and Q. Sun, Phys. Rev. B 69, 045105 (2004).
O. I. Sumbaev, Sov. Phys. JETP 30, 927 (1969).
A. A. Petrunin, A. E. Sovestnov, A. V. Tyunis, and E. V. Fomin, Tech. Phys. Lett. 35, 73 (2009).
O. I. Sumbaev, Sov. Phys. Usp. 21, 141 (1978).
A. E. Sovestnov, A. V. Tyunis, E. V. Fomin, A. A. Petrunin, A. I. Kurbakov, and B. T. Melekh, Pis’ma Zh. Tekh. Fiz. 54, 55 (2012).
A. V. Ivanyuta, A. E. Sovestnov, and E. V. Fomin, Phys. Solid State 54, 688 (2012).
A. E. Sovestnov, A. A. Naberezhnov, Yu. A. Kumzerov, A. A. Sysoeva, V. A. Ganzha, A. I. Egorov, N. M. Okuneva, V. I. Fedorov, and E. V. Fomin, Phys. Solid State 55, 837 (2013).
A. A. Naberezhnov, A. A. Petrunin, A. E. Sovestnov, D. A. Kurdyukov, E. V. Fomin, and S. B. Vakhrushev, Tech. Phys. Lett. 41, 1205 (2015).
I. V. Golosovsky, I. Mirebeau, G. André, D. A. Kurdyukov, Yu. A. Kumzerov, and S. B. Vakhrushev, Phys. Rev. Lett. 86, 5783 (2001).
I. V. Golosovskii, A. A. Naberezhnov, D. A. Kurdyukov, I. Mirebeau, and G. André, Crystallogr. Rep. 56, 164 (2011).
O. I. Sumbaev, in Modern Physics in Chemistry, Ed. by E. Fluck and V. L. Goldanskii (Academic, London, 1976).
A. Kara and T. S. Rahman, Phys. Rev. Lett. 81, 1453 (1998).
S. Hirai, A. M. dos Santos, M. C. Shapiro, J. J. Molaison, N. Pradhan, M. Guthrie, C. A. Tulk, I. R. Fisher, and W. L. Mao, Phys. Rev. B 87, 014417 (2013).
J. Strempfer, U. Rütt, S. P. Bayrakci, Th. Brückel, and W. Jauch, Phys. Rev. B 69, 014417 (2004).
S. Limandri, S. Ceppi, G. Tirao, G. Stutz, C. G. Sanchez, and J. A. Riveros, Chem. Phys. 367, 93 (2010).
S. D. Gamblina and D. S. Urchb, J. Electron Spectrosc. Rel. Phenom. 113, 179 (2001).
V. R. Galakhov, M. C. Falub, K. Kuepper, and M. Neumann, J. Struct. Chem. 49, S54 (2008).
Funding
A.V. Fokin is grateful to the Russian Foundation for Basic Research (grant 19-02-00760). A.A. Naberezhnov is grateful to BRICS-t framework program 19-52-80019 of the Russian Foundation for Basic Research for partial financial support. The development of the synthesis methods of the oxide nanoparticles was performed using the state budget funds as part of state task 0040-2019-0012.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Boltukhina
Rights and permissions
About this article

Cite this article
Naberezhnov, A.A., Sovestnov, A.E., Kurdyukov, D.A. et al. The Effect of Ultradispersion on the Chemical Shifts of the X-Ray K Lines in Copper and Manganese Oxides. Phys. Solid State 62, 1836–1844 (2020). https://doi.org/10.1134/S1063783420100224
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063783420100224