
Abstract
The results of studying the thermally stimulated depolarization currents in thin films of polydiphenylenephthalide (PDP) with a thickness of 20 nm to 1.4 μm under a constant stress in a structure with an insulating dielectric layer are presented. Nonmonotonic behavior of the dependence of the activation energy of charge carrier trapping sites and the relaxation of the space charge on the thickness of PDP polymer films is found. A correlation between the change in the supramolecular structure of the films and the above parameters is established. The obtained results are interpreted using the theory of thermally stimulated discharge in short-circuit heterogeneous structures.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 INTRODUCTION
The high demand for materials of smart electronics has led to the synthesis of a wide range of polymers with unique functional properties. Entire classes of polymers that are capable of changing their electrophysical characteristics under small external stimuli (changes in pressure, light, humidity, atmospheric composition, electric and magnetic fields, radiation, etc.) have been created. These materials include heat-resistant nonconjugated polymers of the class of polyarylenephthalides (PAPs) [1, 2]. Submicron PAP films exhibit abnormally high levels of electrical conductivity comparable to the electrical conductivity of metals. The explanation of the unique electrophysical properties of thin polymer films is associated with the mechanism of delocalization of deep-lying electronic states in the forbidden zone of the polymer when the concentration of the excess charge reaches a certain value [1]. At the same time, the complex spectrum of electronic states in the band gap is a feature of this class of polymers [3–5]. As a rule, the nature of the distribution of these states is determined by the chemical structure of a particular polymer [6]. As was found in [7, 8], the supramolecular ordering in thin films of polydiphenylenephthalide (PDP)—which is a representative of PAPs—changes with a change in the film thickness. It was shown later [9, 10] that some electronic properties of thin films change in the transition region of thicknesses. This does not contradict, in principle, the general idea of the relationship between the structure and properties of polymeric materials. For example, studies of the relaxation processes occurring in R-BAPS polyimide films [11] showed that not only the chemical structure of the polymer, but also the supramolecular structure of the studied objects has an effect on the activation energy of charge carrier trapping sites [12]. A similar trend can be traced in other electronic properties of various polymers [13, 14].
As was found in [7, 8], the supramolecular ordering of PDP polymer films in the thickness range from 100 to 200 nm changes from a homogeneous globular structure to structures with relatively large aggregates of macromolecules. The nature of these changes may be important in predicting the electronic properties of various heterostructures based on wide-gap polymers. However, the relationship between the supramolecular structure of PDP films and the change in the energy distribution of charge traps has not yet been studied.
Therefore, the aim of this work was to study the dependence of the energy characteristics of traps (activation energy) on the thickness of PDP polymer films by the method of thermally stimulated current spectroscopy [15].
2 MATERIALS AND METHODS
Thin films of polydiphenylenephthalide are chosen as an object of this study, since a study of trap states by thermostimulated methods has been performed earlier on such objects [3–6, 16, 17]. In addition, the supramolecular structure of thin films was studied using various methods. The experimental samples were formed on a glass substrate and represented a multilayer metal/polymer/metal system. The lower aluminum electrode was formed by thermal diffusion sputtering. The second copper electrode was of a pressure type. The pressure of the electrode on the sample was chosen on the basis of the following two considerations: it should be less than the threshold pressure of electronic switching of the film [1] and should provide, at the same time, reliable electrical contact. The quality of the contact was evaluated by the type and reproducibility of the current–voltage characteristics (CVC). A polymer film was deposited on a metal electrode by centrifugation (3000 rpm) from a solution of PDP in cyclohexanone. The thickness of the polymer films was set by changing the concentration of solutions in the range from 0.5 to 10 wt %. This corresponded to film thicknesses from 20 nm to 1.4 μm. Samples were dried for 30 min under normal conditions and then in a drying oven for 40 min at a temperature of 150°C to remove solvent residues. The quality and thickness of the polymer films were controlled by atomic force microscopy using an SMM2000T microscope. The dependence of the thickness of the polymer films on the concentration of the polymer solution and the mean square surface roughness of the polymer films are shown in Fig. 1.

(a) Dependence of the polymer film thickness on solution concentration c [8] and (b) mean square roughness Rq of submicron films.
The method of thermally stimulated depolarization (TSD) currents was chosen as a main research method. This method is referred to as highly sensitive methods for studying electronic states in polymeric materials. It is used to study the electrical relaxation mechanisms and energy parameters of electrically active charge-trap sites in dielectrics and semiconductors [15]. Earlier, the study of the spectra of TSD currents in micrometer PDP films [16, 17] revealed some mechanisms responsible for the thermal relaxation of trapping sites. It was found that the main charge relaxation mechanisms are dipole polarization associated with the thermal motion of the lateral phthalide fragments and a change in the concentration of the space charge due to the capture of charges injected from the electrodes by electron traps.
The difficulty in implementing this method is associated with the possible appearance of charge instability in PDP thin films, which leads to switching of the sample conductivity to a high-conductivity state. As was found earlier [1, 18–20], submicron PDP films placed between two metal electrodes can change, at relatively small applied voltages, their conductivity by several orders of magnitude (up to the conductivity of metals). It is obvious that a possible increase in the conductivity of thin polymer films due to injection of charges from the electrodes in an electric field can substantially limit the applicability of the thermally stimulated method. Therefore, a heterogeneous two-layer structure with an insulating dielectric layer was created by analogy with [15] to eliminate the appearance of the equilibrium conductivity of ultrathin polymer films. With this approach, the insulating layer should exhibit low conductivity and have a greater thickness in comparison with the studied film.
Such a structure enables accumulation of charges due to the Maxwell–Wagner polarization at the polymer–insulator interfaces and in the bulk of a thin polymer film. In this case, the expression for thermally stimulated depolarization current j(T) in a two-layer heterostructure will have, taking into account the conductivity of the layers, the following form [15]:
where ε1 and ε2 are the dielectric constants of the polymer film and insulating layer, respectively; d1 and d2 are their thicknesses; Ub is the voltage of the polarizing field; λ1, Ea1, and ω1 are the conductivity, activation energy, and frequency factor of the polymer film, respectively; and T and T1 are the current and initial temperatures, respectively.
As can be concluded from an analysis of dependence (1), the shape of the spectrum of TSD currents is determined by the parameters of the studied polymer film—in our case, the PDP film—rather than by the properties of the insulating layer.
Therefore, a thin film of fluoroplastic F-4 with a thickness of d2 = 10 μm was chosen as an insulating layer located between the polymer film and the upper pressure electrode.
To confirm above statement (1), control measurements of the TSD currents of the specified fluoroplastic film and two-layer film comprised of fluoroplastic and PDP were carried out (Fig. 2). It was found that under specified conditions of measurements, no specific features were recorded in the TSD spectrum of the fluoroplastic (Fig. 2). In this case, the two-layer film exhibits a spectrum that is quite typical for PDP [17]. A comparative analysis of the obtained experimental data confirms the conclusions arising from equation (1). In particular, the fluoroplastic film with a thickness of d2 that significantly exceeds a thickness of d1 of the PDP film and with a conductivity that is lower than the conductivity of PDP has no effect on the position of the temperature maxima and the shape of the TSD spectra. The volume resistivities of the fl-uoroplastic and PDP are about 1017 Ω cm and 1012 Ω cm, respectively [21].

Spectrum of TSD currents for (1) fluoroplastic and (2) PDP films with a thickness of 20 nm in a structure with an insulating fluoroplastic layer. The heating rate is 3 K/min.
Activation energies Ea were estimated using the method of temperature peak position [9]. To calculate the activation energy from the position of the temperature peak, the following formula is applied:
where k is the Boltzmann constant, Im is the maximum current of the corresponding peak in the TSD spectrum, Tm is the temperature of the maximum of the corresponding TSD current peak, σ(Tm) is the charge released during relaxation, and β is the rate of heating.
3 RESULTS AND DISCUSSION
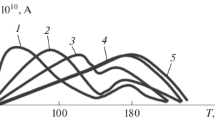
The spectra of TSD currents for PDP films (thicknesses from 20 nm to 1.4 μm) with an insulating fluoroplastic layer are shown in Fig. 3.

Spectrum of TSD currents for PDP films in a structure with an insulating fluoroplastic layer. The temperature scan rate is 3 K/min.
For each spectrum of TSD currents, the following two regions are distinguished: the low-temperature region (from 230 to 270 K) and the high-temperature region (from 270 to 310 K). According to [6, 17], the first region corresponds to dipole polarization, and the second region corresponds to the formation of a charge captured in traps in the polymer film bulk.
Figure 4 shows the experimentally obtained dependences of the temperature position of the peaks and the current maximum on the polymer film thickness for the first and second temperature regions distinguished in the TSD spectra. As is known [15], the amplitude of the peaks of the thermally stimulated depolarization current is usually proportional to the concentration of charge carriers captured in the trap upon charging the dielectric, and the charge value is directly proportional to the voltage of the applied field and the capacitance of the sample. However, complex dependences of the TSD current peak magnitudes both on the thickness of the polymer film and on the temperature position of the depolarization current maxima are observed in the experiment up to film thicknesses of about 200 nm. With an increase in the polymer film thickness from 200 nm and higher, Tm is linearized and shifts to lower temperatures.

Dependences of (a) the current maximum and (b) the temperature position of the peaks on the polymer film thickness for the first (1) and second (2) temperature ranges of the TSD spectra.
The relationship between the temperature position of the TSD current maximum (Tm) and the parameters of trapping sites and the experimental conditions is determined from equation (1) by an extremum analysis, as follows
Thus, the dependence of the temperature peak position on the thickness of the polymer film can be affected by both a change in the energy spectrum of charge trapping sites and an effective frequency factor.
Applying the well-known approximate formulas used in [16], the TSD current maximum can be determined, as follows:
where A are the constant parameters that do not depend on the properties of the studied polymer film. Using relation (4), the dependence of the experimental data on the thickness of the polymer film can be estimated, as follows:
With a weak (insubstantial) dependence of the energy parameters on the film thickness, relation \({{I}_{m}}T_{m}^{2}\) = Bd1 + C should be fulfilled, where B characterizes the change in the electrophysical and energy parameters of the PDP film, and C is a coefficient.
Figure 5 shows the experimental data, which are linearized by two curves with different slopes, within the error margin. The transition of the slope occurs in the region of thicknesses close to 200 nm. According to [7, 8], the supramolecular structure of PDP films changes in this thickness range. Thus, it can be argued that a change in the supramolecular structure of thin films has a substantial effect on the energy parameters.

Dependence of the \({{I}_{m}}T_{m}^{2}\) value on the thickness of the PDP film for the first (1) and second (2) temperature ranges of the TSD spectra.
Figure 6 shows the dependence of the activation energies of trapping sites on the thickness of the polymer film, which is calculated from the experimental data by formula (2). The shape of the activation energy curve corresponds to a nonlinear dependence. Three segments can be distinguished in the curve.

Dependence of the activation energy on the polymer film thickness for the first (1) and second (2) temperature ranges of the TSD spectra.
The first range of thicknesses extends up to 60 nm, in which an increase in the activation energies for both regions is observed. Moreover, the activation energy value for the first region is greater than for the second region.
In the second and third thickness ranges, the activation energy for the second region is greater than for the first region. Moreover, a decrease in the activation energies is observed for thicknesses of up to 200 nm. With a further increase in the thickness of the polymer film, the activation energies increase and the difference in the activation energies from the case of dipole polarization becomes more pronounced (significant).
The results of experimental studies of the supramolecular structure of polymer layers in submicron polydiphenylenephthalide films by the atomic force microscopy and small-angle neutron scattering methods are presented in [7, 8]. It was established that polymer films form, depending on the change in the rheological properties of the solution used in their manufacture, the following two types of supramolecular structure in the bulk of the material: a globular structure and an ellipsoid-shaped structure in the form of associates. The transition from one supramolecular structure to another one is shown in Fig. 6.
The first supramolecular organization corresponds to thicknesses up to 60 nm (Fig. 6). This range of thicknesses corresponds to the minimum roughness of polymer films, the manifestation of homogeneous spontaneous surface polarization, and the reversible total polarization switching initiated by an external field [9, 10]. The observed phenomena correspond to the first segment in the dependence of the activation energy on the polymer film thickness. The large value of the activation energy of dipole polarization relative to the release of space charge is probably associated with the orienting effect of the substrate surface on the ultrathin layers of the polymer film.
The second range, i.e., a segment of the curve in the range of thicknesses from 60 to 200 nm (Fig. 6) in the dependence of the activation energy on the thickness, is associated with the rearrangement of the supramolecular structure of the polymer film, which leads to an increase in the defectiveness of the film structure and a decrease in the activation energies. All this leads to a decrease in the influence of the substrate on dipole polarization and to the appearance of smaller electron traps for the bound space charge.
In the third range of thicknesses (Fig. 6), the activation energies increase. This region corresponds to the formation of associates of macromolecules in the bulk of the polymer film. The appearance of macromolecule associates leads to a change in the polarization conditions and, accordingly, affects the process of thermal relaxation of polarized molecules. At the same time, the second (high-temperature) range of activation energies corresponding to the process of formation of a space charge trapped by electron traps increases sharply, with a tendency to saturation of the activation energy within the limits of 1.2 ± 0.1 eV.
4 CONCLUSIONS
There is a point of view [16] that the activation energy weakly depends on the thickness of the polymer film. In this regard, the presence of three segments in the dependence of the activation energy on the thickness (Fig. 6), which vary within 1.5 eV in the following ranges: from 0.17 to 0.88 eV for the first region and from 0.3 to 1.3 eV for the second region in the selected range of thicknesses. In this case, these segments correlate with the results of structural studies of polymer films [7, 8]. The method of thermally stimulated depolarization currents used in this study has made it possible to reveal a region of critical thicknesses up to 200 nm, which require a more detailed study. It is possible that new properties may be discovered in this region of polymer film thicknesses.
REFERENCES
A. N. Lachinov and N. V. Vorob’eva, Phys. Usp. 49, 1223 (2006).
S. N. Salazkin, V. V. Shaposhnikova, L. N. Machulenko, N. G. Gileva, V. A. Kraikin, and A. N. Lachinov, Polymer Sci., Ser. A 50, 243 (2008).
A. S. Nakaryakov, V. A. Antipin, A. N. Lachinov, D. A. Mamykin, and A. F. Ponomarev, Khim. Vys. E-nerg. 46, 503 (2012).
A. S. Nakaryakov, A. N. Lachinov, A. F. Ponomarev, E. E. Tseplin, and V. A. Antipin, Phys. Solid State 53, 2525 (2011).
V. Kh. Il’yasov, A. N. Lachinov, A. V. Moshelev, and A. F. Ponomarev, Phys. Solid State 50, 568 (2008).
A. F. Ponomarev, A. V. Moshelev, V. Kh. Il’yasov, A. N. Lachinov, V. M. Kornilov, S. N. Salazkin, V. V. Shaposhnikova, D. S. Sharapov, A. O. Burakova, and N. V. Ulitin, Vestn. KTU 17, 159 (2014).
V. M. Kornilov, A. N. Lachinov, D. D. Karamov, I. R. Nabiullin, and Yu. V. Kul’velis, Phys. Solid State 58, 1065 (2016).
D. D. Karamov, V. M. Kornilov, A. N. Lachinov, V. A. Kraikin, and I. A. Ionova, Tech. Phys. 61, 1085 (2016).
R. M. Gadiev, A. N. Lachinov, D. D. Karamov, D. A. Kiselev, and V. M. Kornilov, J. Exp. Theor. Phys. 123, 149 (2016).
D. D. Karamov, D. A. Kiselev, M. D. Malinkovich, V. M. Kornilov, A. N. Lachinov, and R. M. Gadiev, Russ. Microelectron. 45, 619 (2016).
M. E. Borisova, A. L. Didenko, A. M. Kamalov, V. E. Smirnova, and V. E. Yudin, Polymer Sci., Ser. A 60, 751 (2018).
V. A. Zakrevskii and V. A. Pakhotin, Vysokomol. Soedin. A 23, 658 (1981).
B. G. Kim, E. J. Jeong, J. W. Chung, S. Seo, B. Koo, and J. Kim, Nat. Mater. 12, 659 (2013).
E. L. Aleksandrova, M. E. Kompan, M. M. Dudkina, A. V. Tenkovtsev, and E. I. Terukov, Semiconductors 38, 1074 (2004).
Yu. A. Gorokhovatskii and G. A. Bordovskii, Thermal Activation Current Spectroscopy of High-Resistance Semiconductors and Dielectrics (Nauka, Moscow, 1991) [in Russian].
Yu. A. Gorokhovatskii, D. D. Karamov, Yu. Zh. Musralieva, and A. F. Ponomarev, Izv. RGPU im. A.I. Gertsena 157, 60 (2013).
A. N. Lachinov, V. Kh. Il’yasov, and A. F. Ponomarev, Russ. J. Phys. Chem. B 3, 667 (2009).
V. R. Nikitenko, A. R. Tameev, A. V. Vannikov, A. N. Lachinov, and H. Bässler, Appl. Phys. Lett. 92, 138 (2008).
I. R. Nabiullin, A. N. Lachinov, and A. F. Ponomarev, Phys. Solid State 54, 243 (2012).
A. N. Lachinov, V. M. Kornilov, T. G. Zagurenko, and A. Yu. Zherebov, J. Exp. Theor. Phys. 102, 640 (2006).
A. N. Lachinov, A. Yu. Zherebov, and M. G. Zolotukhin, Synth. Met. 59, 377 (1993).
Funding
The study was performed within State assignment no. AAAA-A19-119022290052-9 and supported by grant no. 075-15-2019-1651 for Development of Centers of Collective Use in the Russian Federation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by O. Kadkin
Rights and permissions
About this article

Cite this article
Karamov, D.D., Il’yasov, V.K., Lachinov, A.N. et al. The Effect of Thickness of Submicron Films of Electroactive Polymers on Thermally Stimulated Depolarization Currents. Phys. Solid State 62, 1473–1478 (2020). https://doi.org/10.1134/S1063783420080193
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063783420080193