
Abstract
The features of the composition, crystal, and magnetic structure of Ti-substituted barium hexaferrite BaFe12 – xTixO19 (0.25 ≤ x ≤ 1.5) are studied by Mössbauer spectroscopy, vibration magnetometry, X-ray diffraction, and simultaneous thermal analysis. Bounded heterovalent isomorphism implemented by the scheme 2Fe3+ → Ti4+ + Fe2+ while retaining the charge balance is established when Fe3+ ions are partially substituted with Ti4+ ions. An increase in the 3d-electron density and the presence of Fe2+ in BaFe12 – xTixO19 samples with x = 1.5 is detected by Mössbauer spectroscopy. The heterovalent isomorphic substitution limit is determined to be within 0.75 < x < 1.0. Using Mössbauer spectroscopy and X-ray diffraction, the formation of titanium-contained phases at x = 1.0 whose content increases with the degree of substitution is shown. The data on the preferential distribution of substituent ions (Ti4+) in the barium hexaferrite structure at the 12k and 2b sites are presented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 INTRODUCTION
Magnetic properties of barium hexaferrites depend on both the isomorphic impurity type and content in the structure, which makes it possible of not only to study their properties, but also to synthesize samples with tailored characteristics for practical applications.
M-type hexaferrites and their solid solutions have a crystal structure isomorphic to a natural mineral, i.e., magnetoplumbit PbFe12O19, which was first studied by Adelskold [1].
Iron ions in the hexaferrite with such a structure are localized at five nonequivalent crystallographic sites: 2a, 2b, 4f1, 4f2, and 12k. Among them 2a, 4f2, and 12k are octahedral, 4f1 is tetrahedral, and 2b form a bipyramid. Polyhedra 4f1 and 2a are arranged in the spinel block (S), 4f2 and 2b are in the hexagonal block (R), and 12k is at the interface of spinel and hexagonal blocks (RS) [2].
In unsubstituted hexaferrite BaFe12O19, the collinear magnetic structure is implemented, in which magnetic moments of sites 12k, 2a, and 2b are directed to one side, while 4f1 and 4f2 are directed in antiparallel to the other side [3], which results in uncompensated antiferromagnetism (ferrimagnetism). Weakening of these interactions due to the substitution of Fe3+ ions of both groups with nonmagnetic and/or low-magnetic metal ions results in a decrease in the resulting magnetic moment and can lead to a noncollinear magnetic structure [4].
To controllably change magnetic and electrical properties, the principle of a change in the hexaferrite chemical composition is used.
In this case, most researchers considered the isovalent substitution of Fe3+ ions with metal ions with close ionic radius and an oxidation state of 3+, since the charge balance is implemented rather easily in this case (electrical neutrality law conservation) [5, 6]. The introduction of two or several various elements of the same valence also did not cause problems with charge balance, but complicates interpretation of their localization in the structure [7]. A much more complex situation arises for the charge balance if we consider heterovalent substitutions. Some authors, to provide the charge balance in the synthesis, use substitutions of Fe3+ ions with a Ме2+ + Ме4+ pair. Substitutions with Ti4+ simultaneously with divalent ions Mn2+, Со2+, Zn, Ni were studied in many works. For example, in [8], Fe3+ ions in strontium hexaferrite are substituted with Mn2+ + Ti4+ ions with the general crystallochemical formula SrFe12 – 2xMn\(_{x}^{{2 + }}\)Ti\(_{x}^{{4 + }}\)O19. Although according to [8], Mn ions are localized at the site 4 f1, there are different opinions on Ti4+ localization. A large number of studies are devoted to the study of barium hexaferrites alloyed with Ti4+ ions simultaneously with Со2+ ions, e.g., [9]. The results obtained show that the substitution of Fe3+ ions with Ti4+ ions simultaneously with Co2+ ions do not lead to a change in the hexaferrite structure, the charge balance in this case is retained, but the magnetization of samples decreases. In [9], it is believed that such materials can be used as radio absorbing ones.
Similar results leading to a decrease in the magnetizations as Ti4+ and Mg2+ ion pair is introduced into the structure are presented in [10] with an indicated increase in the unit cell parameter with the degree of substitution x. In the case of Fe3+ ion substitution with Ti4+ + Zn2+, a higher isomorphic capacity of hexaferrites and the dependence of the saturation magnetization on methods and conditions of their synthesis are indicated in [11]. In [12], experiments on the introduction of three elements (Ti4+, Mn2+, Cu2+) at x = 1, 2, and 3 into the lattice were performed; according to X-ray diffraction, hexaferrite was still single-phase at x = 2. However, the appearance of the second phase at x = 2 was detected in [13]. A decrease in the saturation magnetization and an increase in the permeability with x was indicated. An increase in the number of substituting ions to four resulted in ambiguity of results. In [14], it is believed that the obtained material with no agglomeration is applicable to high-frequency magnetic devices. In [15], even at x = 0.5, magnetic characteristics featured complex behavior as a result of the ambiguous cationic distribution of substituting elements in the structure. It should be noted that it is rather difficult to determine the cation localization at a large variety substituting ions; furthermore, it is difficult to determine the domination of any substituting ion on hexaferrite properties.
Since isovalent substitutions of Fe3+ ions with single elements and Ме2+ + Ме4+ pair are widely enough studied, it is of interest to consider heterovalent substitutions of Fe3+ ions separately with Me2+ and Me4+ ions. Few sources suggest that the isomorphic capacity of hexaferrite in the case of titanium introduction is slightly higher than for other elements, which extends the applicability of such hexaferrites in microwave techniques. Taking this into account, hexaferrite alloyed with Ti4+ ions was chosen for study.
Among early studies of titanium-alloyed barium hexaferrites is [16], in which a natural hexaferrite analogue of composition BaFe10Ti2O19 found in Germany and approved as a new mineral Batiferrit was studied. Its composition, physical properties, and structure, which corresponded to the magnetoplumbit structure according to X-ray diffraction, were studied. In [17], the study of hexaferrites with x = 0, 0.6, 0.8, and 1.0 by X-ray diffraction and magnetometry showed a change in the unit cell parameters and titanium introduction into tetrahedral (17%) and octahedral (75%) sites, which leads to a decrease in the remanent magnetization and magnetic anisotropy with increasing Ti content. In this case, it is believed that the magnetization increases at low titanium concentrations and decreases with increasing titanium concentration.
A series of M-type barium hexaferrites was synthesized with partial substitution of Fe2O3 with TiO2 in [18]. The melt composition (mol %) was 40BaO + 33B2O3 + (27 – x)Fe2O3 + xTiO2, where x = 0, 3.6, 5.4, and 7.2 mol % of TiO2.
Substituted ferrites were studied using X-ray diffraction, Mössbauer spectroscopy, and magnetometry. The X-ray diffractions data showed that all samples, except for the ferrite phase, had a BaTi6O13 phase impurity. Mössbauer spectra of compositions with x = 0, 3.6, 5.4 mol % of TiO2 showed magnetic ordering with strong line broadening. Furthermore, line broadening increased with the TiO2 content. According to Mössbauer parameters, Ti4+ ions occupy sites 2a and 12k; at high contents, the also occupy sites 2b and 4 f2, which causes a decrease in the coercive force and saturation magnetization.
Single crystals of Ti-substituted barium hexaferrite BaFe12 – xTixO19 with x < 1.3 were studied in [19, 20]. The Ti distribution over various crystallographic sites was determined by the X-ray diffraction data. At low Ti contents (x to 0.8), the unit cell expands; as x further increases, the unit cell begin to constrict. This is associated with the Fe3+-to-Fe2+ transition and vacancy formation to maintain the charge balance. In this case, the Curie temperature and magnetization monotonically decrease.
Literature sources show that there is no consensus on the cationic distribution in the hexaferrite structure and in particular on titanium localization. In some works, it is stated that Ti4+ ions can enter almost all sites except for 4 f1 [18]; in others, its incorporation into site 2b is denied [17]. Questions also arise on the charge balance mechanism in hexaferrite when Ti4+ ions enter the lattice and the appearance of ions with an increased isomer shift to 0.49 mm/s, related immediately to Fe2+ ions [18], and the formation of additional phases, depending on the titanium concentration.
Therefore, the objective of this work is to study the correlation of chemical and phase composition and magnetic properties as functions of the preferential distribution of the heterovalent substituent ion in the BaFe12 – xTixO19 samples under study with 0.25 ≤ x ≤ 1.5 in the implementation of the charge balance scheme.
2 OBJECTS AND METHODS OF STUDY
The objects of study were polycrystalline barium hexaferrite ВаFe12 – хTixO19 samples, where x = 0.25, 0.5, 0.75, 1.0, 1.25, and 1.5. The samples were produced by the ceramic technology from ultrapure grade oxides Fe2O3, TiO2, and carbonate ВаСО3. The preliminary initial composition was subjected to synthesizing annealing in air at 1200°C (6 h) and then was sintered at 1300°C (6 h). After sintering, samples were slowly cooled in a furnace (~100°/h) [13].
The Mössbauer spectra of BaFe12 – xTixO19 samples were measured using an Ms-1104 Em spectrometer with constant acceleration, the number of points of 512, a Со57 γ-radiation source in a chromium matrix, at room temperature. The isomer (chemical) shift was calculated with respect to α-Fe. Powder samples 0.05–0.07 mm in size made of sintered ferrite were used. The mathematical treatment of the spectra was performed by the Univem Ms program.
The magnetic parameters, i.e., the specific magnetization σs, remanent magnetization σr, coercive force Hc, and hysteresis loop shape, were measured using a VSM 250 vibration magnetometer in a magnetic field of 20 kOe at 300 K.
The Curie temperature of samples was determined using a СТА449С simultaneous thermal analysis setup in a magnetic field. The phase composition of samples was determined using a Rigaku Ultima IV diffractometer with Bragg–Brentano focusing, based on CuKα radiation with a diffracted-beam graphite monochromator. Point-by-point scanning mode was used; the 2θ angular range was 20°—140°, the step was 0.05. The unit cell parameters of hexaferrites were calculated by interplane distances.
3 RESULTS AND DISCUSSION
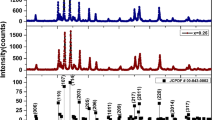
The Mössbauer spectra of hexaferrite ВаFe12 – xTiхO19 samples are shown in Fig. 1. All spectra were expanded into sextets and doublets using the Univem Ms program. The expansion model was set from the following considerations. In substituted ferrites, sextets belonging to iron ions of main five sites with parameters of unsubstituted hexaferrite were separated, and positions of additional sextets and doublets were visually determined. The best processing version was determined by the minχ2 parameter while retaining the physical meaning of Mössbauer parameters. Decreases in total intensities of sextet peak positions were used to determine structure sites occupied by impurity ions and broken magnetic couplings of the indirect Fe–O–Fe magnetic exchange. In this case, it was taken into account that nonequivalent sites of Fe ions in the case of substituting ion sites 2a, 2b, 4f1, and 4f2 can be formed in the neighboring polyhedra with which exchange magnetic couplings are broken, and only impurity ion incorporation into site 12k can form nonequivalent sites 12k' due to joining three octahedra 12k.

Mössbauer spectra of M-type hexagonal ferrites BaFe12 – xTixO19 (x = 0.25, 0.5, 0.75, 1.00, 1.25, 1.50).
According to these considerations, the Mössbauer spectra of samples with x = 0.25 and 0.50 were expanded to 6 sextets, and the spectrum of the sample with x = 0.75 was expanded to 7 sextets. An additional doublet appeared in the spectrum of the sample with x = 1.0; the spectra of samples with x = 1.25 and 1.5 were expanded to 8 sextets and a doublet. Such an expansion provided the best cases of the Pearson criterion min χ2. In this case, an increase in x was accompanied not only by an increase in the number of sextets in experimental spectra, but also an increase in the total intensity of doublets. The obtained parameters of sextets and doublets, i.e., the isomer (chemical) shift δ (mm/s), quadruple splitting Δ (mm/s), the magnetic field at Fe57 nuclei, spectrum component areas S (rel %), resonance line width Γ (mm/s), their correspondence with occupied sites, and the angle θ between the sample magnetic moment and γ-radiation vector in the spectrometer are listed in Table 1. For comparison, Table 1 shows the parameters of one of unsubstituted barium hexaferrites. Since the main contribution to additional sextets C6 and C7 is made by nonequivalent ions of site 12k, they are denoted by 12k' and 12k".
The appearance of the doublet in the spectrum of the sample with x = 1, on the one hand, suggests that titanium ceased incorporation into the hexaferrite ВаFe12 – хTixO19 structure, and its saturation reached; on the other hand, we can see solid solution decomposition and the formation of new iron–titanium phases. Since a doublet is not still observed in hexaferrite with x = 0.75, whereas its total intensity is significant at x = 1, it can be considered that the limit of bounded isomorphism of ВаFe12 – хTixO19 hexaferrite is in the range 0.75 < x < 1.0. Furthermore, an antiferromagnetic α-Fe2O3 phase is detected in Mössbauer spectra with x = 1.25 and x = 1.5.
Estimating the Mössbauer parameters of Table 1, we can note variations of the Fe3+ ion isomer shift δ of five basic sites (sextets 1–5) for both unsubstituted and substituted hexaferrites. Comparisons show high δ for Fe3+ ions of octahedral sites 12k, 4f2, and 2a (δ = 0.33–0.39 mm/s) in comparison with tetrahedral 4f1 and bipiramidal 2b sites (δ = 0.23–0.29 mm/s) according to [21]. This is explained by the larger covalence of bonds between iron ions due to the smaller volume of polyhedra 4f1 and 2b. In this case, the quadruple splitting Δ of Fe ions increases due to a larger deviation of octahedron 12k symmetry from the ideal one. In this case, only ligands contribute to the electric field gradient [22, 23]. In this regard, noteworthy are significant distortions of polyhedron 2b symmetry (Δ = 2.07–2.23 mm/s), whereas Δ varies within 0.1–0.18 mm/s for the most symmetric octahedron 2a.
A significant increase in δ additional sextets can be explained, on the one hand, by a decrease in the bond covalence of Fe3+ ions, when octahedron 12k donates iron ions for the formation of iron–titanium structures and the formation of cation vacancies in this case. This was accompanied by an increase in interaction distances in the triad of octahedra 12k, which results in strengthening the ionic bond. In this case, the density of P-electrons decreases due to an increase in the screening effect of d-electrons with the result of increasing isomer shift.
At the same time, of interest is the implementation scheme of the charge balance in hexaferrite with isomorphic titanium. The classical substitution scheme 2Fe3+ → Ti4+ + Fe2+ involves divalent iron which should be recorded using Mössbauer spectroscopy. Ions completely satisfying the Fe2+ state in the range 0.25 ≤ x ≤ 0.75 were not detected; however, a small increase in δ of Fe sextets C6 (12k') with respect to C1 (12k) of unsubstituted hexaferrite takes place. A significant increase in δ is observed in additional sextets at x → 1.0. For example, in the sample with x = 1.0, sextet С6 with δ = 0.44 mm/s, appears in the spectrum; at x = 1.25, sextets С6 with δ = 0.50 mm/s and С7 with δ = 0.56 mm/s appear; and at x = 1.5, sextet with δ = 0.63 mm/s appears (Table 1).
It cannot be considered that formed ions completely satisfy the Fe2+ state, since, as shown in the study [21] on systematization of valence and coordination states of Fe2+ ions, δ for Fe2+ in the environment of O2– should be within 0.94 ± 0.02 mm/s for tetrahedral sites and 1.14 ± 0.02 mm/s for octahedral sites. However, if we assume that the electron exchange Fe3+ → Fe2+ occurs in the spinel block of hexaferrite in octahedra 12k as that in magnetite established in [24], the isomer shift for Fe ions of octahedral sites of magnetite in Mössbauer spectra should be 0.67 mm/s which was established in numerous studies of magnetite. In the sample with x = 1.5, the isomer shift of Fe3+ ions is 0.65 mm/s, which is relatively close to 0.67 mm/s as in unsubstituted magnetite during electron exchange.
From this we can conclude that only a partial electron exchange occurs with increasing 3d-electron density at small substitutions of Fe3+ with Ti4+ ions in the spinel block of hexaferrite, whereas the electron exchange mechanism Fe3+ ↔ Fe2+ similar to that in magnetite is implemented at x > 1.0. The relatively close coincidence of the isomer shift in additional sextets at x = 1.5 with that observed in magnetite for octahedral sites points to the high probability of the explanation of an increase in the isomer shift by the electron exchange at the 12k site of hexaferrite, rather than the model of strengthening the ionic bond, which cannot provide the observed increase in the isomer shift in additional sextets of the Mössbauer spectrum.
In contrast to magnetite, where Δ of Fe ions is zero during electron exchange due to the high octahedron symmetry, in hexaferrite BaFe12 – xTixO19, due to the defect structure formation, local distortions of the octahedron are rather strong, and Δ reaches 0.5 mm/s, which is unrelated to electron exchange.
Magnetic fields at Fe57 nuclei monotonically decrease with increasing x. Comparing sextet areas, a certain deviation of areas in tetrahedral sites in the bipyramid from theoretical values can be noted (see Table 1). It can be explained by different vibration amplitudes of Fe ions in these polyhedra.
The smaller vibration amplitude of Fe ions in tetrahedra in comparison with octahedra enhances the resonant effect, hence, increases the area from these ions, and the large amplitude of Fe ion vibrations in the bipyramid in one of the directions decreases the resonant effect.
A comparison of the angle θ of BaFe12 – xTixO19 samples with x confirms that all samples are almost isotropic.
The measured magnetic characteristics of the samples studied in the x range from 0 to 0.75 of the specific magnetization M, remanent magnetization Mr, coercive force Hc, and Curie temperature TC in the form of dependences on the degree of substitution x are shown in Fig. 2. The specific magnetization (Fig. 2a) increases from 68 emu/g of unsubstituted hexaferrite to 69 emu/g at x = 0.25, and then monotonically decreases to 62 emu/g at x = 0.75. This results from the fact that the substitution at the initial stage occurs mostly at the site 4 f2, increasing the resulting magnetic moment, and then substitutions 12k and 2b prevail (see Table 1), which decrease the resulting magnetic moment, hence, the specific magnetization, which corresponds to the data [17]. The coercive force and remanent magnetization behave identically, decreasing from the values of unsubstituted hexaferrite to the values at x = 0.25, and then they remain almost unchanged to x = 0.75. The Curie temperature measured using simultaneous thermal analysis of hexaferrites in a magnetic field (Fig. 2d) showed a decreasing linear dependence.

Dependences of BaFe12 – xTixO19 magnetic characteristics on x: (a) specific magnetization Ms, emu/g; (b) coercive force Hc, kOe; (c) remanent magnetization Mr, emu/g; and (d) Curie temperatures TC, °C.
The field dependences of the magnetization (Fig. 3a) and scaled-up fragments (Fig. 3b) for unsubstituted hexaferrite and compositions with x to 0.75 showed their certain difference. It may be noted that complete magnetization saturation was not reached in the measured range of B(T); we can see two groups of hysteresis loops: loops with higher (x = 0, 0.25, 0.5) and lower (x = 0.75) specific magnetizations. Furthermore, hysteresis loops of the sample with x = 0.25 are closer to rectangularity, which is consistent with the magnetization peak (Fig. 2a).

(a) Hysteresis loops of hexaferrite BaFe12 – xTixO19; (b) the scaled-up portion with B = 1–3.
Using a simultaneous thermal analysis, thermal characteristics of samples with x = 0–0.75 were measured; the dependences of differential scanning calorimetry (DSC) and mass loss (thermogravimetry, TG) in a magnetic field obtained at the same time are shown in Fig. 4. Endothermic effects associated with the sample transition from the magnetically ordered ferrimagnetic state to the paramagnetic state are indicated in all curves; therewith, the transition temperature decreases strictly linearly from 452.4 to 371.3°C. The same dependence is reflected by the TG curve measured in a magnetic field (Fig. 2d). In this case, during the transition of the magnetically ordered sample at the Curie temperature, a signal step appears in the temperature dependence of TG, by which the transition temperature can be determined. To compare the effect of the magnetic field on the TG signal, the sample with x = 0.25 was measured without magnetic field, where the transition signal in the TG curve is absent. Graphically, the signal in the TG curve is less smeared than the endothermic peak; therefore, the determination of the Curie temperature is more accurate in this case.

Curves of differential scanning calorimetry (DSC) and mass loss (thermogravimetry, TG) of BaFe12 – xTixO19 hexaferrites (x = 0, 0.25, 0.50, 0.75).
The phase composition of samples with x = 0–0.75, 1.25, and 1.5 was determined by X-ray diffraction patterns (Fig. 5); the unit cell parameters of hexaferrites calculated by interplane distances are listed in Table 2.

X-ray diffraction patterns of hexaferrites BaFe12 – xTixO19 (x = 0, 0.25, 0.50, 0.75, 1.00, 1.50).
We can see in Table 2 that samples in the composition range x = 0–0.75 inclusive represent a BaFe12 ‒ xTixO19 monofractions.
This suggests that titanium isomorphically enters the hexaferrite lattice within this range. Beginning with x = 1.0, the X-ray diffraction data establish the appearance of various iron–titanium compounds (Table 2) at an almost unchanged composition of hexaferrite BaFe11.25Ti0.75O19. This indicates bounded isomorphism of hexaferrite and isomorphism limit in the range 0.75 < x < 1.0, which is consistent with the Mössbauer spectroscopy data showing the absence of a doublet in the spectra of samples with x = 0.75 and the doublet appearance at x = 1.0.
The calculated BaFe12 – xTixO19 unit cell parameters (Table 2) show that the titanium incorporation into the lattice decreases the parameter a in the composition with x = 0.25 with respect to unsubstituted hexaferrite, but a further increase in x does not affect this parameter. In contrast to the parameter a, the parameter c and unit cell volume V show maxima in the range of 0.75–1.0, which can also be associated with reaching the isomorphism limit.
4 CONCLUSIONS
The performed study showed that bounded heterovalent isomorphism according to the scheme 2Fe3+ → Ti4+ + Fe2+ takes place in titanium-alloyed barium hexaferrite. It was found that the limit of heterovalent isomorphic substitution in BaFe12 – xTixO19 is in the range of 0.75 < x < 1.0; then, titanium in compound with iron, separated from sites 12k and 2b, forms paramagnetic ulveshpinel, magnetite, and titanium dioxide according to the X-ray diffraction and Mössbauer spectroscopy data. In samples with x = 1.25 and 1.5, iron oxide α-Fe2O3 was determined by Mössbauer parameters. The formation of iron–titanium compounds and titanium dioxide is detected at x = 1.0, and their content increases with x. The electron exchange Fe3+ ↔ Fe2+ in the range x = 1.0–1.5 in the spinel block of the BaFe12 – xTixO19 structure was detected by the isomer shift similar to that observed in magnetite. Additional sextets in Mössbauer spectra of barium hexaferrite can be formed in the case of Ti4+ ion localized mostly at sites 12k and 2b, due to broken exchange couplings Fe(12k)–O–Fe(12k), Fe(4 f2)–O–Fe(12k), Fe(2a)–O–Fe(12k), and Fe(4 f2)–O–Fe(2b) denoted by 12k' and 12k".
The dependence of the specific magnetization of BaFe12 – xTixO19 exhibited a maximum at x = 0.25 due to the prevalence of the substitution at the site 4 f2, after which the magnetization monotonically decreases with increasing x.
The studies performed show the application ranges of titanium-substituted barium hexaferrite and the possibility of obtaining samples with prescribed magnetic properties for industrial applications.
REFERENCES
V. Adelskold, Ark. Mineral. Geol. A 12, 1 (1938).
R. C. Pullar, Prog. Mater. Sci. 57, 1191 (2012).
J. Smit and H. P. J. Wijn, Ferrites (Phylips Tech. Lib., 1959).
V. V. Korovushkin, M. N. Shipko, V. G. Kostishin, I. M. Isaev, A. Yu. Mironovich, S. V. Trukhanov, and A. V. Trukhanov, Inorg. Mater. 55, 1007 (2019).
A. S. Kamzin and L. P. Ol’khovik, Phys. Solid State 41, 1658 (1999).
V. V. Korovushkin, A. V. Trukhanov, M. N. Shipko, V. G. Kostishin, I. M. Isaev, A. Yu. Mironovich, and S. V. Trukhanov, Russ. J. Inorg. Chem. 64, 574 (2019).
A. V. Trukhanov, V. G. Kostishin, V. V. Korovushkin, L. V. Panina, S. V. Trukhanov, V. A. Turchenko, I. S. Po-lyakov, R. Kh. Rakhmatulin, G. A. Filatov, T. I. Zu-bar’, V. V. Oleinik, E. C. Yakovenko, L. Yu. Matsui, L. L. Vovchenko, V. L. Launets, and E. L. Rukhanova, Phys. Solid State 60, 1768 (2018).
Sh. Sh. Bashkirov, A. V. Liberman, A. A. Valiulin, L. D. Zaripova, and S. V. Kokin, Phys. Solid State 42, 79 (2000).
T. Tsutaoka, A. Tsurunaga, and N. Koga, J. Magn. Magn. Mater. 399, 64 (2016).
M. H. Shams, A. Rozatian, M. Yousefi, J. Valıček, and V. Šepelák, J. Magn. Magn. Mater. 399, 10 (2016).
V. V. Somana, V. M. Nanotib, D. K. Kulkarnic, and V. V. Somand, Phys. Proc. 54, 30 (2014).
A. Ghasemi, A. Hossienpour, A. Morisako, A. Saatchi, and M. Salehi, J. Magn. Magn. Mater. 302, 429 (2006).
R. Alam, M. Tehrani, M. Moradi, E. Hosseinpour, and A. Sharbati, J. Magn. Magn. Mater. 323, 1040 (2011).
Y. Zheng, Z. Yu, Y. Shao, S. Mo, and Y. Lin, Hyperfine Interact. 94, 2035 (1994).
R. Alam, M. Moradi, H. Nikmanesh, J. Ventura, and M. Rostami, J. Magn. Magn. Mater. 402, 20 (2016).
C. L. Lengauer, E. Tillmans, and G. Hentschel, Mineral. Petrol. 71, 1 (2001).
P. A. Mariño-Castellanos, J. Anglada-Rivera, A. Cruz-Fuentes, and R. Lora-Serrano, J. Magn. Magn. Mater. 280, 214 (2004).
P. Quoiroz, B. Halbedel, A. Bustamante, and J. Gonzalez, Hyperfine Interact. 202, 97 (2011).
D. A. Vinnik, D. A. Zherebtsov, L. S. Mashkovtseva, A. S. Semisalova, I. V. Krivtsov, L. I. Isaenko, G. G. Mikhailov, and R. Niewa, Cryst. Growth Des. 14, 5834 (2014).
D. A. Vinnik, D. A. Zherebtsov, L. S. Mashkovtseva, N. S. Perov, A. S. Semisalova, I. V. Krivtsov, L. I. Isaenko, and G. G. Mikhailov, in Proceedings of the 6th Baikal International Conference,2014, p. 161.
F. Menil, J. Phys. Chem. Solids 46, 763 (1985).
R. Ingalls, Phys. Rev. A 133, 787 (1964).
G. Bancroft, A. Maddock, and R. Burns, Geochim. Cosmochim. Acta 31, 2219 (1967).
E. J. W. Verwey and P. H. Haaman, Physica (Amsterdam, Neth.) 8, 979 (1941).
Funding
This study was supported by the Russian Science Foundation, agreement no. 19-19-00694, May 6, 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by A. Kazantsev
Rights and permissions
About this article

Cite this article
Korovushkin, V.V., Trukhanov, A.V., Kostishin, V.G. et al. Study of Features of the Composition, Magnetic, and Crystal Structure of Barium Hexaferrite BaFe12 – xTixO19. Phys. Solid State 62, 891–901 (2020). https://doi.org/10.1134/S1063783420050145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063783420050145