
Abstract
Two derivatives of adamantane benzenesulfamide have been synthesized and studied: N-adamantane-1-yl-2,4,6-trimethyl-benzenesulfamide (1) and N-adamantane-1-yl-4-methoxy-benzenesulfamide (2). Crystal structures 1 and 2 are solved using X-ray diffraction (XRD) analysis. The solubilities of compounds in a phosphate buffer solution with pH 7.4, 1-octanol, and n-hexane are determined at several temperature points using isothermal saturation. The values of Gibbs energy, enthalpy, and entropy of solubility of the compounds at 298 K are determined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Currently, sulfanilamide derivatives are widespread drugs with a wide range of therapeutic action: antibacterial, hypoglycemic, and diuretic [1, 2]. In recent years, anticancer activity of some sulfanilamide-containing compounds has been revealed [3].
In many respects, the efficiency of potential drugs is determined by not only their structure but also the possibility of being transferred in an organism to the disease site. The water solubility of compounds is considered as the basic factor; however, one should not ignore the lipophilicity of compounds, which determines the ability of molecules to penetrate cellular membranes. Most of tested potential medicinals are usually characterized by only one solubility type (liophilicity or lipophilicity). Introduction of additional fragments into potential biologically active compounds often solves the aforementioned problem.
In recent years, adamantane fragments have been used to modify developed medicinal forms [4, 5]. Adamantane has the following useful properties due to its spherical structure: high degree of lipophilicity, chemical stability, thermal stability, conformational hardness, etc. Adamantane substituents, which exhibit high lipophilicity and practically have no toxicologic effects, facilitate the ability of drugs to penetrate cellular membranes and thus improve their pharmacokinetics.
Molecules containing substituents in the adamantane fragment have a complex spatial structure; their solubility and other characteristics change in dependence of the substituent nature and may have different degrees of biological activity.
This study is a continuation of the investigations devoted to the synthesis and analysis of the structure and physicochemical characteristics (including solubility in lyophilic and lipophilic media) of adamantane sulfanilamide derivatives [6–8].
EXPERIMENTAL
To continue the search for efficient potential medicinal forms and study the influence of molecular design on the crystal structure and solubility of compounds in lyophilic and lypophilic media, the following two adamantane benzenesulfanilamide derivatives were synthesized and studied according to the techniques described previously in [8]:

The structure of the compounds was confirmed using 1H NMR spectroscopy (Bruker CXP-200 spectrometer, operating frequency 200 MHz, CDCl3 solvent, tetramethylsilane as an internal standard) and elemental analysis (Carlo-Erba CHN analyzer).
N-Adamantane-1-yl-2,4,6-Trimethyl-Benzenesulfanilamide (1). The results of elemental analysis are as follows: (C) 68.49, (H) 8.33, and (N) 4.11 wt %. The chemical formula is C19H27NO2S. The calculated contents are (C) 68.43, (H) 8.16, and (N) 4.20 wt %. The 1H NMR spectrum (chemical shift δ): 1.57 (m, 6H, AdH), 1.76 (m, 6H, AdH), 1.98 (broadened s, 3H, AdH), 2.28 (s, 3H, CH3), 2.65 (s, 6H, 2CH3), 4.40 (s, 1H, NH), and 6.92 ppm (s, 2H, ArH).
N-Adamantane-1-yl-4-Methoxy-Benzenesulfanilamide (2). The results of elemental analysis are as follows: (C) 63.32, (H) 7.28, and (N) 4.52 wt %. The chemical formula is C17H23NO3S. The calculated contents are (C) 63.52, (H) 7.21, and (N) 4.36 wt %. The 1H NMR spectrum (δ): 1.55 (m, 6H, AdH), 1.76 (m, 6H, AdH), 1.98 (broadened s, 3H, AdH), 3.85 (s, 3H, OCH3), 4.63 (broadened s, 1H, NH), 6.93 (d, J = 8.80 Hz, 2H, ArH), and 7.82 ppm (d, J = 8.80 Hz, 2H, ArH).
Single crystals of compounds 1 and 2 were grown from 96% ethanol by slow evaporation. Materials were completely dissolved in solvent upon weak heating and continuous stirring. The solutions obtained were slowly evaporated at room temperature through small holes in a protective film covering weighing bottles with solutions.
Crystallographic characteristics, details of the X-ray experiment, and refinement parameters for structures 1 and 2 are listed in Table 1. The structures were directly solved [9, 10] according to the program [11] and refined using full-matrix least-squares method in the anisotropic approximation of displacements of all atoms except for hydrogen. The hydrogen atoms near nitrogen atoms were localized based on the Fourier difference maps, and their positional and thermal parameters were refined in the isotropic approximation. Other hydrogen atoms were placed in geometric sites and involved in refinement in correspondence with the rider model. The calculations were carried out using the SHELXTL program [12]. The crystallographic data were deposited with the Cambridge Crystallographic Data Centre (CCDC nos. 1900204 and 1900202); they are freely accessible at www.ccdc.cam.ac.uk/data_request/cif.
The solubility of compounds 1 and 2 was studied in 1-octanol (purity more than 99%, Merk, Germany) and n-hexane (of analytical grade, Khimmed, Russia), both used without further purification, and in a phosphate buffer solution. The buffer aqueous phosphate solution with pH 7.4 was prepared by mixing solutions of phosphate acid potassium and sodium salts (KH2PO4 and Na2HPO4 of analytical grade, Reakhim, Russia) according to the technique described in [13]. The salt concentrations were chosen so that the ionic strength of the obtained solutions corresponded to 0.15 mol/L (physiological value). The рН was measured using a pH-meter-430 (Corning).
The solubility was determined experimentally using isothermal saturation at several temperature points (20, 25, 30 (or 32), 35 (or 37), and 42 (or 45) ± 0.1°C). All experimental data are presented in molar fractions. Solid phase was removed by filtering through Acrodisc CR syringe filters (PTFE, pore diameter 0.2 µm) or by centrifugation in a Biofuge Pico centrifuge (Kendo Laboratory, Germany) at a rotational speed of 10000 rpm for 3 min. All filters, pipettes, syringes, and glassware that contacted initial solutions were previously thermostated at experimental temperatures. The experimental results are presented as averaged values based on at least three experiments. The molar solubility of the compounds was measured on a spectrophotometer with an error of 2–2.5% using the protocol reported in [14].
The standard values of the Gibbs energy of dissolution processes, \(\Delta G_{{{\text{sol}}}}^{0}\) [kJ mol–1], were calculated from the equation
where X2 is the molar fraction of material in a saturated solution at 298.15 K and γ2 is the solute activity coefficient.
The standard dissolution enthalpy \(\Delta H_{{{\text{sol}}}}^{0}\) [kJ mol–1] was calculated according to the Van’t Hoff equation
assuming that the activity coefficient γ2 is unity and the dissolution enthalpy is independent of concentration because of the low solubility of the materials under study.
The temperature dependences of the solubilities of materials within the chosen temperature range can be described by the linear function
where A is an integral coefficient related to entropy and B = \(\Delta H_{{{\text{sol}}}}^{0}\)/R.
The values of standard dissolution entropy \(\Delta S_{{{\text{sol}}}}^{0}\) [J mol–1 K–1] were obtained from the Gibbs equation:
RESULTS AND DISCUSSION
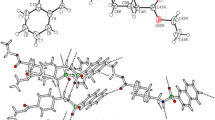
According to the XRD data, the structural units of compounds 1 and 2 are centrosymmetric dimer associates N-adamantane-1-yl-2,4,6-trimethyl-benzenesulfanilamide (Fig. 1) and molecules of N-adamantane-1-yl-4-methoxy-benzenesulfanilamide (Fig. 2). It can be seen that the presence of two methyl substituents in the benzene ring of molecules barely affects their conformation. The torsion angles between the benzene ring and adamantane fragments are 86.20(13)° and 62.7(3)° in molecules 1 and 2, respectively. The bond lengths in the adamantane, sulfanilamide, and benzene fragments are standard and correspond to the values determined previously for related structures [5, 6]. In a crystal, molecules 1 and 2 are stacked according to the “head-to-head” type and oriented along the b axis. The parameters of the intermolecular hydrogen bonds, which combine molecules 1 and 2 into dimer associates, are listed in Table 2. There are no hydrogen bonds between the dimer associates in stacks.

Centrosymmetric dimer associate formed by molecules 1.

Centrosymmetric dimer associate formed by molecules 2.
Since compounds 1 and 2 have a potential biological activity, it is of interest to study their dissolubility in physiological liquids and their lipophilic properties, which facilitate penetration of cellular membranes. A phosphate buffer solution with рН 7.4, 1-octanol, and n-hexane were chosen as model solvents. Most biochemical reactions occur in strictly defined pH ranges. Blood plasma with рН 7.4 is an important medium for passive transport. 1-Octanol solvent is convenient for simulating a lipid layer of a cellular membrane. The n-hexane/buffer system is used to simulate a hematoencephalic barrier: n-hexane (involved in only nonspecific interactions) imitates a nonpolar region of a brain, whereas the buffer imitates the blood system.
The energy aspects of interaction of compounds 1 and 2 with solvents were estimated by calculating the thermodynamic functions of processes. The temperature dependences of the solubility of compounds in the aforementioned solvents are presented in Table 3, and the thermodynamic solubility functions are given in Tables 4 and 5.
As one can see in Table 3, introduction of a methoxyl group into a para-position of the benzene ring (compound 2) increases its solubility in the buffer solution in comparison with compound 1, which contains three methyl groups in the molecular benzene fragment. The standard values of enthalpy and entropy of dissolution of compound 2 in the phosphate buffer solution are much larger than those for compound 1.
The solubility of compound 2 in 1-octanol also exceeds that of compound 1; however, the thermodynamic dissolution functions change only slightly (the values are consistent within the experimental error). At the same time, the solubility of compound 2 in n-hexane is lower than that of compound 1. The standard enthalpy and entropy of dissolution of compound 2 exceed the corresponding values calculated for compound 1; although the calculated values differ not so much as for the solubility in the phosphate buffer solution. On the whole, the data obtained correspond to the previous results of studying the dissolution processes for many substituted adamantane benzenesulfamides [8].
REFERENCES
J. Drews, Science 287 (5460), 1960 (2000). https://doi.org/10.1126/science.287.5460-5464.1960
C. T. Supuran and A. Scozzafava, Expert Opin. Ther. Pat. 10 (5), 575 (2000). https://doi.org/10.1517/13543776.10.5.575
F. Abbate, A. Casini, T. Owa, et al., Bioorg. Med. Chem. Lett. 14 (1), 217 (2004). https://doi.org/10.1016/j.bmcl.2003.09.062
G. L. Perlovich and T. V. Volkova, Phys. Chem. Chem. Phys. 20 (30), 19784 (2018). https://doi.org/10.1039/c8cp03716g
A. P. Voronin, T. V. Volkova, A. B. Ilyukhin, et al., CrystEngComm 20 (25), 3476 (2018). https://doi.org/10.1039/c8ce00426a
G. L. Perlovich, A. M. Ryzhakov, N. N. Strakhova, et al., J. Chem. Thermodyn. 69 (1), 56 (2014). https://doi.org/10.1016/j.jct.2013.09.027
G. L. Perlovich, A. M. Ryzhakov, V. V. Tkachev, and A. N. Proshin, CrystEngComm 17 (25), 753 (2015).
G. L. Perlovich, T. V. Volkova, A. V. Sharapova, et al., Phys. Chem. Chem. Phys. 18 (13), 9281 (2016). https://doi.org/10.1039/c6cp00379f
G. M. Sheldrick, Acta Crystallogr. A 64 (1), 112 (2008). https://doi.org/10.1107/S0108767307043930
G. M. Sheldrick, Acta Crystallogr. A 71 (1), 3 (2015). https://doi.org/10.1107/S2053229614024218
G. M. Sheldrik, SHELX-86, Program for the Crystal Structure Determination (Univ. of Cambridge, Cambridge, England, 1986).
G. M. Sheldrick, SHELXTL v. 6.14, Structure Determination Software Suite (Brucker AXS, Madison, Wisconsin, USA, 2000).
G. L. Perlovich, T. V. Volkova, A. N. Proshin, et al., Mol. Pharm. 8 (5), 1807 (2011). https://doi.org/10.1021/mp2003237
G. L. Perlovich and A. Bauer-Brandl, Curr. Drug Deliv. 1 (3), 213 (2004). https://doi.org/10.1002/jps.20611
Funding
This study was supported by the Ministry of Science and Higher Education of the Russian Federation (project no. 075-03-2020-223 (FSSF-2020-0017)) and within the State assignment (state registration no. 0089-2019-0011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by Yu. Sin’kov
Rights and permissions
About this article

Cite this article
Kovalchukova, O.V., Kazachenko, V.P., Tkachev, V.V. et al. Crystal Structure and Physicochemical Characteristics of Two Novel Adamantane Derivatives of Sulfonamides. Crystallogr. Rep. 66, 424–428 (2021). https://doi.org/10.1134/S1063774521030123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1063774521030123