
Abstract
We used a plastid ndhC-trnV intergenic spacer to reconstruct the genealogy of haplotypes and phylogenetic relationships of major taxonomic groups of the genus Rosa. We analyzed the data using the statistical parsimony, maximum likelihood, and NeighborNet approaches. According to our results, the majority of species may be subdivided into three groups roughly corresponding to the taxonomic sections Pimpinellifoliae, Rosa (former Cinnamomeae) and Synstylae + Chinenses + Gallicanae + Caninae. We assess their relationships as broadly paraphyletic, since the first group appears to be ancestral to the second and the third. The fourth group distanced from these three by many mutational steps consists of R. persica (subgenus Hulthemia) sequences. We argue that R. persica together with the North American R. minutifolia and R. stellata (section Minutifoliae) probably represent the only remnants of an ancient group of roses widely distributed in the Northern Hemisphere in the Oligocene. Two major lineages of the section Caninae arose independently from different ancestors belonging to Synstylae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The genus Rosa comprises about 150 to 500 species (Buzunova, 2001; Ku and Robertson, 2003; Wissemann, 2003) distributed in temperate areas of the Northern Hemisphere. The exact number of wild species is still debated among taxonomists. An unusual difficulty with the genus Rosa, for systematic classification and delimitation of species, was first mentioned by C. Linnaeus who singled it out with a special commentary in the first edition of his Species Plantarum (Linnaeus, 1753): “Species Rosarum difficile distingvuntur, difficilius determinantur; mihi videtur naturam miscuisse plures vel lusu ex uno plures formasse; hinc qui paucas vidit species facilius eas distinguit, quam qui plures examinavit” (The species of the genus Rosa are difficult to distinguish and determine; I have seen that nature may mix up many of them or, just for fun, forms a new one out of the lot; those who have seen only a few distinguish them more easily than those who have examined a lot).
Wissemann (2006) and Tomljenović and Pejić (2018) presented reviews of the history and current state of taxonomic knowledge of the genus, including several molecular phylogenetic studies and their impact on taxonomy. To sum up the current state of the art, the World Checklist of Vascular Plants (Govaerts, 2022) lists 367 names of species and hybrids as accepted, and 663 as synonyms. Another database, Plants of the World Online (2022), lists 271 accepted species and hybrid names among wild roses.
The contemporary classification of the genus Rosa still relies on works from the 19th and 20th centuries despite quite a number of molecular phylogenetic studies produced in recent years. The latest classification by Wissemann (2003) is actually based on Rehder’s classification (Rehder, 1949), which in turn is based on taxonomic concepts developed by Crépin (1869, 1891).
According to Wissemann (2003), the genus Rosa is subdivided into four subgenera, the largest subgenus Rosa being comprised of ten sections. In modern taxonomic treatments (Ku and Robertson, 2003; Lewis et al., 2022) taking into account the results of recent molecular genetic studies (see below), the genus Rosa may be treated as comprised of two to four subgenera with ten to 12 sections within the subgenus Rosa. Actually, however, only two subgenera are accepted: Hulthemia with a single species R. persica, and Rosa, to which all the other species belong. After conservation of the genus name Rosa with the lectotype of R. cinnamomea (International Code…, 2006; International Code…, 2018), application of the autonymous sectional name Rosa has changed: the section Rosa became Gallicanae, while the section Cinnamomeae had its name changed to Rosa.
Molecular studies of the 21st century have resulted in contradictory phylogenetic schemes, compared in detail in Koopman et al. (2008) and Fougère-Danezan et al. (2015). All these phylogenetic trees are either poorly resolved, or, if some resolution was achieved, have low support of inner nodes. Only recent studies using nuclear single-copy gene tags (Debray et al., 2019) or complete plastome sequences (Cui et al., 2022) enabled researchers to reconstruct trees with good support of basal and internal nodes. Unfortunately, these works were not aimed at the genus phylogenetic reconstructions as such, but at the assessment of the usefulness of the corresponding markers to reconstruct the reticulate evolution of Rosa, in the first case, and at revealing the origin of a number of hybrid cultivars, in the second. Hence, both of these works employed inadequate and unbalanced taxon samples (Supplement, Table S1). Nevertheless, although phylogenetic trees reconstructed by different authors are based on different sets of taxa and different markers (Supplement, Table S1), they possess several common topological patterns, taxonomic interpretation of which has been generally accepted. Thus, the North American species traditionally referred to as the section Carolinae appeared to belong to the section Rosa (former Cinnamomeae) clade in all the studies.
Another finding confirmed by most phylogenetic studies is that the genus Rosa can be roughly subdivided into two major clades, one mostly including species of the Rosa and Pimpinellifolae sections, the other one uniting mostly members of the Synstylae and Caninae sections (Wissemann and Ritz, 2005; Bruneau et al., 2007; Fougère-Danezan et al., 2015; Zhu et al., 2015; Liu et al., 2015; Debray et al., 2019; Cui et al., 2022; Zhang et al., 2022). Members of the sections Chinenses, Bracteatae, Laevigatae, and Microphyllae are usually associated with this second clade.
When many samples of the section Caninae are involved in the analyses, they either form a separate clade or split into two to three sister clades together with the west Eurasian species of the Synstylae section (Wissemann and Ritz, 2005; Fougère-Danezan et al., 2015). Phylogenetic studies also indicate isolated basal positions of R. persica (subgenus Hulthemia) and R. minutifolia and R. stellata (section Minutifoliae) which sometimes cluster together on a long branch (Wissemann and Ritz, 2005; Fougère-Danezan et al., 2015; Zhang et al., 2022). Rosa roxburghii (section Microphyllae), R. laevigata (section Laevigatae), and R. banksiae and R. cymosa (section Banksianae) form one to several separate clades sister to the clade of the Synstylae section (Fougère-Danezan et al., 2015; Zhu et al., 2015; Zhang et al., 2022), thus confirming their sectional taxonomic status assumed on morphological grounds.
Due to the different sets of taxa used in the studies, it is often difficult to assess the exact position and relationships of certain species. The problem is even worse, because many species, especially in the sections Caninae and Rosa, cannot be reliably determined by their morphological characters, and various authors might use genetically different samples under the same names. Many species of Rosa can hybridize when growing together in nature and in botanical gardens and nurseries. This additionally complicates the problem of comparability of different phylogenetic trees due to the possible hybrid origins of samples.
Another common characteristic of the trees obtained by various authors is that many terminal and intermediate interior branches are very short or even zero long, while at least some of the deeper interior branches are long. This indicates the recent origin of most of the species and clades, while a few of them are old and distantly related to the others. This result was recently corroborated by Zhang et al. (2022) with the analyses of the complete plastomes of 37 species of Rosa. These authors concluded that members of the subgenus Rosa underwent a rapid simultaneous diversification, which seems to be the reason for short and poorly supported branches in phylogenetic trees. They also identified species of the section Minutifoliae (R. stellata and R. minutifolia) as the earliest diverging clade and those of the subgenus Hulthemia (R. persica, under the name R. berberifolia) and the section Pimpinellifoliae (represented in their work by R. xanthina and R. omeiensis) as its successive clades.
All the above difficulties with interpretation of phylogenetic trees result in continuing discussion of the evolution of roses in terms of their outdated classifications, attempting to tie the revealed clades with traditional sectional subdivisions of the genus based on morphology.
Phylogenetic analysis algorithms may face problems when data do not represent a tree-like structure (Jacob and Blattner, 2006). In the abovementioned study, the authors analyzed the phylogeny of the genus Hordeum and demonstrated that deep coalescence (Hudson, 1990) can result in incongruence between nuclear and plastid phylogenetic trees both in young, rapidly speciating groups, and in old lineages reaching deep into the history of the genus. To overcome these problems, they reconstructed the genealogical relationships of plastid haplotypes using the statistical parsimony approach for the whole genus. The same approach was used successfully to reconstruct the phylogeny and biogeography of the Allium subgenus Melanocrommium (Gurushidze et al., 2010) and Chinese species of Gagea (Peterson et al., 2019). This is probably also the case for Rosa, where extensive hybridization between various species coupled with shallow ancestry of many of them, and in combination with several old isolated lineages, creates a reticular evolutionary pattern that cannot be reflected adequately by phylogenetic trees. In such a situation we suggest analysis of genealogical relationships of the plastid haplotypes instead of a species phylogenetic tree using the statistical parsimony network approach. Plastid haplotypes genealogy mostly reflects the maternal lineage of ancestry, which is less prone to the influence of reticular evolution. According to the present knowledge, plastids are inherited strictly maternally in roses (Bruneau et al., 2007), though evidence of heteroplasmy in rose hybrids has appeared recently (Schanzer et al., 2020). Nevertheless, tracing plastid haplotype genealogies may be more successful in assessment of the limits and relationships of major groups of roses than the tree based phylogenetic reconstructions. This approach to the phylogenetic analysis of recently diverged groups was justified by Jacob and Blattner (2006) and its use suggested for Rosa by Schanzer (2011). We have chosen a plastid intergenic spacer (IGS) ndhC-trnV to trace the maternal lineage of roses and to minimize the effects of reticular evolution on phylogenetic reconstruction to reveal the limits and relationships of major groups within the genus. This region was shown as highly variable for different groups of flowering plants (Shaw et al., 2007), including the genus Rosa (Fedorova et al., 2010; Meng et al., 2011; Schanzer et al., 2011, 2020; Zhu et al., 2015; Zhang et al., 2022). The use of a single marker is often criticized as insufficient for phylogeny reconstruction. However, in our case several multi-marker phylogenetic trees, based on both nuclear and plastid sequences, had already been published, but this did not help to get better resolution or support of the basal nodes of the trees. In our study we test the potential of the haplotype network approach to reconstruct the evolution of the genus Rosa. We need to stress that our approach was intended to reconstruct the genealogy of Rosa plastid haplotypes but not the Rosa species phylogeny per se. However, haplotypes or groups of closely related haplotypes may be indicative of species or groups of species relationships. In our study, we tried to include several individuals from different origins for each species. To check our results achieved from a single plastid marker for consistency, we also analyzed a small sample of full plastid genome sequences available from GenBank using the maximum likelihood (ML) approach and NeighborNet algorithm and compared the resulting trees and split graphs with the haplotype network.
MATERIALS AND METHODS
Plant Material
Plant material for this study was almost completely collected in the wild from Western Europe (Germany, Check Republic, Hungary, Croatia, Italy), Ukraine, the European part of Russia, Siberia, the Russian Far East, the Caucasus, and Uzbekistan. Since we expected incomplete lineage sorting among closely related recently diverged groups of species, we sampled several individuals per species whenever possible, only 37 species were represented by singular sequences (Supplement Table S2). We determined voucher specimens to species level using identification keys in several Floras (Klášterský, 1968; Buzunova, 2001; Henker, 2003; Ku and Robertson, 2003) and comparing our specimens with type materials kept in Herbaria of St. Petersburg (LE), Kyiv (KW), Lvov (LW), Vienna (W), and Berlin (B), when available. Because the ndhC-trnV IGS was also used in a phylogenetic study of the sections Synstylae and Chinenses (Zhu et al., 2015), which were undersampled in our collection, we used the sequences from this study available in GenBank (Zhu et al., 2015) to include in our data matrix (Supplementary Table S2). From the sequences available, we selected only those obtained from specimens collected in the wild, except for several sequences of local species cultivated in botanical gardens. We also used several ndhC-trnV sequences from taxa with complete plastid genomes available in GenBank (Jian et al., 2018a, 2018b; Wang, Q., et al., 2018; Chen, X. et al., 2019; Chen, M. et al., 2019; Jeon, et al., 2019; Meng, et al., 2019; Wang, M., et al., 2019; Zhang, C. et al., 2019; Zhang, S.D. et al., 2019; Zhao, et al., 2019; Cui, et al., 2020; Yin, et al., 2020). Altogether the data matrix of ndhC-trnV sequences comprised 363 sequences obtained from our specimens and 140 sequences obtained from GenBank, representing 101 species from all sections except for the North American section Minutifoliae (formerly subgenus Hesperhodos) with two species, R. minutifolia and R. stellata.
DNA Extraction, PCR, and Sequencing
Total genomic DNA was extracted from silica gel dried leaves using the NucleoSpin Plant II DNA extraction kit (Macherey-Nagel, Germany) according to the manufacturer’s instructions. The ndhC-trnV region was amplified with ndhC (ATTAGAAATGYCCARAAAATATCAT) and trnV(UAC)x2 (GTCTACGGTTCGARTCCGTA) primers (Shaw et al., 2007). Primers used for PCR were synthesized and purified in PAAG by Syntol Ltd. (Moscow, Russia). Polymerase chain reaction (PCR) was performed in a total volume of 20 µL, containing 4 µL of Ready-to-Use PCR MasterMIX based on “hot-start” SmarTaq DNA Polymerase (Dialat Ltd., Moscow, Russia), 13 µL of deionized water, 3.2 pmol of each primer, and about 1.5–2 ng of template DNA in a MJ Research PTC-220 DNA Engine Dyad Thermal Cycler (BioRad Laboratories, United States) under the following conditions: 95°C–180 s; 95°C–60 s, 57°C–40 s, 60°C–80 s (35 cycles); 57°C–40 s, 60°C–80 s (2 cycles). Amplification products were checked in 1% agarose gel in 0.5× TBE buffer with ethidium bromide staining and purified by precipitation in 0.125M/L ammonia acetate solution in 70% ethanol. Purified PCR products were sequenced in both directions using the ABI PRISM © BigDye™ Terminator v. 3. kit (Applied Biosystems) and further analyzed on an ABI PRISM 3730 automated sequencer (Applied Biosystems) at the Severtsov Institute of Problems of Ecology and Evolution, at the Genome Common Facilities Centre (Moscow, Russia) and at Syntol Ltd. (Moscow, Russia). All sequences were deposited in GenBank (https://www.ncbi.nlm.nih.gov/); the accession numbers of the sequences are shown in the Supplementary Table S2.
Data Analyses
The sequences ndhC-trnV were aligned using MAFFT v. 7.147b (Katoh et al., 2002, 2013) with the L-INS-i alignment algorithm, followed by manual adjustments in BioEdit (Hall, 1999). Forty-five full plastid genome sequences belonging to 29 Rosa species and ten accessions of outgroup taxa obtained from GenBank were aligned using MAFFT with the FFT-NS-2 alignment algorithm. Since ndhC-trnV alignments had numerous indels, they were checked for uninformative and dubiously aligned regions and trimmed with the BMGE v. 1.12 software (Criscuolo and Gribaldo, 2010). The remaining indels were simple-coded as single mutation events (Simmons and Ochoterena, 2000) with the FastGap v. 1.2 software (Borchsenius, 2009) and appended to the end of the alignment.
The processed alignment was analyzed with the statistical parsimony algorithm described in Templeton et al. (1992) and implemented in the TCS v. 1.21 program (Clement et al., 2000). To find out the probable root of the haplotype network obtained with the TCS, we prepared a haplotype alignment, containing only one sequence per haplotype, and a species alignment, containing one to several sequences per species, depending on the number of haplotypes revealed within species. These alignments were analyzed with an outgroup using the maximum likelihood approach in the raxmlGUI 2.0 beta (Silvestro and Michalak, 2012; Edler et al., 2019), and NeighborNet algorithm as implemented in the SplitsTree4 (Huson, 1998; Huson and Bryant, 2006) programs. A separate analysis was performed using the maximum likelihood approach on a sample of 45 full plastid genome sequences obtained from GenBank. All the analyses in the raxmlGUI 2.0 were performed with the default settings. The program searched for trees with the maximum likelihood approach under the GTRGAMMA model with parameters calculated by the program. Bootstrap values are based on 100 replicates (Fast bootstrap option).
To root the network, we used a multiple outgroup including sequences of Rubus occidentalis (OK054548), R. arcticus (OL891648), R. pedunculosus (OQ992654), R. biflorus (OQ992653), R. coreanus (MH992398), R. trifidus (MK465682), Hagenia abyssinica (KX008604), Sanguisorba officinalis (MF678801), S. tenuifolia (MH513641), and Potentilla micropetala (KY420021). We chose the outgroup taxa according to available phylogenetic trees of Rosaceae (Potter et al., 2002; Eriksson et al., 2003; Zhang et al., 2017).
RESULTS
ndhC-trnV IGS Statistical Parsimony Analysis
Sequences of Rosa truncated from both ends varied from 383 to 475 positions long. The resulting ndhC-trnV alignment was 588 positions long. After coding gaps as single mutational events and trimming with BMGE, the final alignment was 583 positions long. Statistical parsimony analysis in TCS collapsed sequences into 95 haplotypes and calculated the maximum number of connection steps with more than 95% confidence of parsimony as ten steps (parsimony limit). All the haplotypes were united into a single network. We failed to analyze the alignment with the outgroup due to considerable excess of the parsimony limit.
The network contains several closed loops caused by homoplasy. We resolved some of the loops using rules described in Crandall and Templeton (1993). Several loops remain unresolved which nevertheless do not hamper discerning relationships among most of the haplotypes (Fig. 1). We follow Crandall and Templeton (1993) in defining tip haplotypes as having a single connection to another haplotype, and interior haplotypes as having two to multiple connections to other haplotypes. Below, we describe the network in terms of haplogroups (groups of closely related haplotypes) I to IX defined basing on their position in the network, and the number and length of connections between the haplotypes included into such a group. Such haplogroups in many cases correspond roughly to the taxonomic sections of the genus Rosa with a number of notable exceptions.

Statistical parsimony network of ndhC-trnV haplotypes of the genus Rosa. Haplogroups are marked with Roman numerals and outlined. The size of circles correlates with the number of accessions representing a haplotype. Black dots correspond to intermediate missing haplotypes absent in the sample. Resolved closed loops are shown with dotted lines. Haplotype colors and legend designations (in the upper left corner) correspond to taxonomic sections of the genus Rosa. Correspondence of haplotype numbers to species and sections is explained below (for more detail, see Supplement Table S2): I: 1–5: subgen. Hulthemia: 1–5 R. persica; II–IX: 6–95: subgen. Rosa: II: 6 + 26–37: sect. Pimpinellifoliae: 6 R. spinosissima; 26, 29–30, 32–34, 36 R. kokanica; 27 R. koreana; 37 R. graciliflora + sect. Rosa: 26 R. macrophylla; 31 R. acicularis; 35 R. prattii; 37 R. moyesii + sect. Microphyllae: 26 R. praelucens + sect. Synstylae: 28 R. glomerata III: 14–25: sect. Pimpinellifoliae: 14–25 R. kokanica IV: 7–13: sect. Rosa: 7 R. caudata, R. davidii; 9 R. multibracteata; 11 R. palustris; 12 R. acicularis, R. nutkana; 13 R. acicularis + sect. Synstylae: 7 R. abyssinica; 8 R. glomerata; 10 R. multiflora, R. multiflora var. cathayensis + sect. Caninae: 12 R. canina, R. stylosa, R. sancti-andreae V: 38–46: sect. Rosa: 38 R. cinnamomea; 39 R. pendulina, R. willmottiae var. glandulifera, R. fedtschenkoana; 40 R. fedtschenkoana; 41 R. amblyotis, R. cinnamomea, R. rugosa, R. davurica; 42 R. davurica; 43–44 R. cinnamomea; 45 R. arkansana, R. woodsii; 46 R. palustris VI: 47–52: sect. Microphyllae: 47, 49 R. roxburghii; + sect. Laevigatae: 48 R. laevigata + sect. Banksianae: 50 R. banksiae; 52 R. cymosa + sect. Pimpinellifoliae: 51 R. omeiensis VII: 53–63: sect. Synstylae: 53 R. deqenensis, R. soulieana, R. soulieana var. sungpanensis; 54 R. derongensis, R. duplicata, R. lichiangensis, R. soulieana; 58 R. anemoniflora, R. brunonii, R. multiflora, R. multiflora var. cathayensis, R. filipes, R. fujisanensis, R. helenae, R. henryi, R. kwangtungensis, R. lasiosepala, R. longicuspis, R. lucieae, R. maximowicziana, R. paniculigera, R. pricei, R. rubus, R. sambucina, R. shangchengensis, R. longicuspis var. sinowilsonii, R. transmorrisonensis, R. uniflorella, R. weisiensis; 59 R. shangchengensis; 60 R. setigera; 62 R. pricei + sect. Chinenses: 56 R. odorata, R. odorata var. gigantea, R. odorata var. pseudoindica; 57 R. odorata var. gigantea; 58 R. chinensis var. spontanea, R. chinensis cult. ‘Old Blush’; 61, 63 R. lucidissima + sect. Synstylae × sect. Rosa: 58 R. × archipelagica VIII: 64–70: sect. Caninae: 64 R. tomentosa, R. villosa, R. rubiginosa, R. elliptica, R. marginata, R. turcica, R. glauca, R. sicula, R. agrestis, R. gremlii, R. hungarica; 65 R. villosa; 66 R. rubiginosa; 67 R. rubiginosa, R. agrestis, R. micrantha; 68 R. × canina, R. pygmaea; 69 R. sicula; 70 R. rubiginosa, R. sicula, R. canina, R. gremlii + sect. Synstylae: 64 R. sempervirens IX: 71–95: sect. Caninae: 72 R. pygmaea, R. corymbifera; 73–75, 78–79 R. pygmaea; 77 R. rubiginosa; 80 R. canina; 81 R. canina, R. pygmaea, R. rubiginosa, R. chomutoviensis; 82 R. pygmaea; 83 R. canina, R. pygmaea, R. rubiginosa, R. chomutoviensis, R. schmalhauseniana, R. subpomifera; 84–85 R. canina; 86 R. marginata; 87 R. pygmaea, R. canina, R. subcanina, R. corymbifera, R. caryophyllacea, R. caesia, R. montana, R. cuneicarpa; 88 R. glauca; 89 R. corymbifera; 90 R. pygmaea; 91–92 R. arabica; 93 R. agrestis; 94–95 R. canina + sect. Gallicanae: 71, 76 R. gallica + sect. Synstylae: 71 R. arvensis, R. phoenicia.
Haplogroup I comprises haplotypes 1 to 5 of R. persica (subgenus Hulthemia), which are distanced one to four mutational steps from each other and by nine mutational steps from the nearest haplotype 6 of the haplogroup II. The haplogroup I has the most isolated position in our network. Haplotype 6 belongs to the haplogroup II and represents sequences of R. spinosissima from two localities in the East Carpathians, and one in France. The haplogroup II comprises mostly haplotypes of the section Pimpinellifoliae with several exceptions. They are R. macrophylla (section Rosa) and R. praelucens (section Microphyllae) sharing interior haplotype 26 with accessions of R. kokanica from several localities in Uzbekistan; R. glomerata (section Synstylae) represented by two accessions sharing tip haplotype 28; R. acicularis, R. prattii, and R. moyesii (section Rosa) represented by tip haplotypes 31, 35, and 37, respectively. The haplogroup II is central in the network being directly or indirectly ancestral to all the other groups.
The haplogroup III also represents the section Pimpinellifoliae and comprises haplotypes 14 to 25 of R. kokanica from a single locality in Uzbekistan.
The haplogroup IV represents haplotypes 7 to 13 mostly belonging to species of the sections Rosa and Synstylae. Only haplotype 12 is shared between samples from the sections Rosa and Caninae. Two haplotypes (11 and 12) of North American members of the section Rosa (R. palustris and R. nutkana) belong to this haplogroup.
The haplogroup V comprises haplotypes 38 to 46 representing all the remaining species of the section Rosa from Eurasia and North America. The haplogroup VI represents a lineage with numerous missing intermediate haplotypes to which haplotypes of the olygotypic sections Microphyllae, Laevigatae, and Banksianae belong. As one can see from Fig. 1, in the initial analysis this lineage shaped a large closed loop caused by homoplasy, resolved using rules in Crandall and Templeton (1993). Haplotype 51 is distanced from this group by six mutational steps and represents a single accession of R. omeiensis (section Pimpinellifoliae). We do not include it in this group because its position varies in other analyses (see below).
The basal haplotype of this haplogroup (47) representing three accessions of R. roxburghii is ancestral to this lineage as well as to haplogroup VII representing most of the accessions of the section Synstylae and all the accessions of the section Chinenses. The majority of accessions of this haplogroup representing different species of both sections share the same internal haplotype 58.
The haplogroup VII and haplotype 58 in particular are ancestral to two independent closely related lineages representing haplogroups VIII and IX. These haplogroups are represented by closely related haplotypes distanced from each other by one mutational step. They comprise the majority of accessions of species of the section Caninae. The haplogroup VIII comprises haplotypes 64 to 70 representing species of the section Caninae (the majority of accessions from the subsections Rubigineae, Rubrifoliae, Trachyphyllae, and Vestitae, with several accessions from the subsection Caninae). The basal interior haplotype 64 is shared with R. sempervirens, a west Eurasian member of the section Synstylae. The haplogroup IX comprises haplotypes 71 to 95 representing the remaining species of the section Caninae (subsection Caninae and a minor part of accessions from the subsections Rubigineae, Rubrifoliae, Trachyphyllae, and Vestitae), as well as species of the section Gallicanae. Its basal interior haplotype 71 comprises the majority of accessions of R. gallica as well as accessions of two west Eurasian species of the section Synstylae, R. phoenicia and R. arvensis.
Geographic Distribution of Haplogroups
Mapping the haplogroup ranges onto a geographical map of the world (with some extrapolations based on previous knowledge of certain taxa ranges) reveals a nonrandom pattern of their distribution (Fig. 2). The range of the haplogroup I (R. persica only) is confined to Central Asia and North Iran reaching Western China in the East. The range of the haplogroup II mostly represented by members of the Pimpinellifoliae section is centered in central China, with its discontinuous parts scattered in northeastern China, the Russian Far East, Central Asia, and Europe. Most of the findings are from Sichuan Province of China. However, its central and most abundantly sampled interior haplotype 26 is almost solely confined to the mountains of Uzbekistan with one sample from Xizang (China). Two other haplotypes were sampled from Inner Mongolia (North China) and south of the Russian Far East. The distribution of the haplogroup III, as has been mentioned above, is restricted to a single locality of R. kokanica in Uzbekistan (Supplement Table S2). The distribution of the haplogroup IV encompasses temperate Eurasia and North America, reaching the Arabian Peninsula and northeastern Africa in the south. It is, however, uneven, with most of the samples originating from southeastern China (Sichuan, Chongqing, Guizhou, and Anhui provinces). Its distribution in North America is not clear with only two samples from the Pacific and Atlantic coasts included in our data set. The haplogroup V is the most widely distributed through nearly all Northern Eurasia and North America. Its range includes several separate parts in Northern Eurasia, with one outlier, the most southerly fragment, in Sichuan Province of China. The distribution of the haplogroup VI is restricted to southeastern China, centered in Yunnan and Sichuan provinces. Only haplotype 49 was sampled from Southern Japan (R. roxburghii var. hirtula). The haplogroup VII is distributed throughout southern and central China, reaching Northern Pakistan in the West, Southern Japan in the East, and the southern part of the Russian Far East in the Northeast. The group has a disjunct range with a fragment in the southeastern part of North America (R. setigera). Haplogroup VIII is confined to Europe, in the southwestern part penetrating the Atlas Mountains in North Africa. The range of the haplogroup IX mostly coincides with it, but is wider, penetrating further to the North, reaching Central Asia in the East, and the Sinai Peninsula in the Southeast.

Geographical distribution of major haplogroups of Rosa. Colors correspond to haplogroup numbers as in Fig. 1 and are shown in the legend in the right upper corner.
NeighborNet Analysis of ndhC-trnV Haplotypes
Since no outgroup could be included in the TCS network of Rosa ndhC-trnV haplotypes due to the excess of the calculated parsimony limit, establishing the basal haplotype was necessary for the network rooting. Analysis of the species alignment in raxmlGUI resulted in a basally unresolved tree with low to zero support of most of the branches (not shown). The analysis of the haplotype alignment in SplitsTree4 using the NeighborNet algorithm resulted in a split graph network where haplotypes 1−5 of R. persica were closest to the outgroup (Fig. 3). This enabled us to root the TCS network on the haplogroup I comprising haplotypes of R. persica (Fig. 1).

NeighborNet network of ndhC-trnV haplotypes of the genus Rosa. Haplotype numbers are explained in Supplement Table S2. The Roman numerals indicate haplogroups as in Fig. 1. The haplogroup I is highlighted as the root of the network. The haplotype 51 (R. omeiensis) is highlighted in bold to indicate its different position compared to the TCS network.
Though this analysis was chiefly performed for rooting purpose, it also revealed roughly the same groups of haplotypes as the statistical parsimony network (Fig. 3). In the split graph, three main groups of haplotypes are resolved. The most early diverged group of haplotypes is shaped by a poorly resolved bunch of edges, corresponding to haplogroups I to V and represented mostly by sequences of species belonging to the sections Pimpinellifolae and Rosa. Among them, haplogroup I, corresponding to the subgenus Hulthemia, appears closer to the outgroup than the others. The haplotype 51 positioned on a long branch connected to the haplogroup VI in the TCS network appeared among the bunch of branches of the haplogroups II and IV in the NeighborNet split graph. The terminal group of branches unites haplogroups VII to IX representing sequences of species belonging to the sections Synstylae, Chinenses, Gallicanae, and Caninae and is in the most distant position from the outgroup. The haplogroup VI representing sequences of species belonging to the sections Microphyllae, Banksianae, and Laevigatae is positioned between them.
Analysis of Full Plastid Genome Sequences
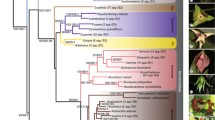
To check our results independently, we analyzed an alignment of 45 full plastid genomes of roses. Full plastid genome sequences of 45 Rosa accessions belonged to 29 species, and ten accessions of outgroup taxa were obtained from GenBank. The sequences of Rosa species varied from 156 333 to 157 391 bp. The aligned sequences were trimmed with BMGE software to delete unreliably aligned parts. The final alignment after trimming was 155 119 positions long. The maximum likelihood analysis in raxmlGUI resulted in a completely resolved and well supported tree (Fig. 4) with a R. persica (subgenus Hulthemia) clade sister to all the other Rosa accessions except for the sequences of R. xanthina, R. sericea, and R. omeiensis. The latter shaped the most early diverged clade, which was represented by only the haplotype 51 of R. omeiensis in our analyses of ndhC-trnV haplotypes. The position of the R. minutifolia MT755634 plastome in the clade uniting members of the sections Rosa and Pimpinellifoliae, and the position of the R. rugosa MN661138 plastome in the clade uniting members of the sections Synstylae and Chinenses, both may be probably explained by wrong taxonomic identification of the initial material used for sequencing (Zhao and Gao, 2020; Yin et al., 2020).

Maximum likelihood tree of Rosa based on full plastome sequences. Colors and Roman numerals to the right indicate haplotype groups and correspond to ndhC-trnV haplogroups in Fig. 1. Bootstrap support values above 50% are shown above branches. Thick branches indicate 100% bootstrap support. Number 51 to the right of the first diverging clade corresponds to the haplotype number of R. omeiensis in haplotype networks in Figs. 1 and 3. Wrong taxonomically identified accessions are highlighted in bold.
DISCUSSION
Rooting the Network
The rooting procedure is crucial for phylogenetic inference, since the topology of a phylogenetic tree depends fully on the position of the root (Kinene et al., 2016). An unrooted tree or network can define separate clades but cannot indicate their evolutionary relationships. In the best resolved phylogenetic tree of Rosaceae (Zhang et al., 2017), Rosa is sister to the Potentilleae clade including Potentilla, Alchemilla, Fragaria, Chamaerhodos, Drymocallis, and Sibbaldia. This clade is, in turn, sister to the clade Agrimonieae. The clade Rubus is sister to all of them. We chose a multiple outgroup and got the clade of R. persica haplotypes to be closest to the outgroup in the NeighborNet analysis of ndhC-trnV haplotypes (Fig. 3). This result was confirmed by plastid genomic analysis of 45 accessions representing the majority of Rosa taxonomic sections (Fig. 4). Our results, as well as other phylogenetic reconstructions based on analyses of full plastomes (Cui et al., 2022; Zhang et al., 2022), revealed the R. persica clade as sister to the majority of other groups of roses, though it was not the earliest divergent. In the study by Zhang et al. (2022), the earliest divergent was the clade R. minutifolia (section Minutifoliae); the next divergent clade, as it appeared in our analysis, was the clade R. xanthina–R. omeiensis, and the next divergent clade was R. persica. This result differs from the reconstruction by Fougère-Danezan et al. (2015), the analyses of the plastid data of which revealed the clade R. xanthina–R. omeiensis as a late divergent and included into their major ‘C and allies’ clade. However, the analysis of various tree topologies obtained using different plastid markers and full plastomes in the above cited papers assures us of the correctness of rooting of our TCS network with the haplogroup I.
Major Taxonomic Groups of Roses
Thus, R. persica and, probably, also the North American R. minutifolia and R. stellata (section Minutifoliae, not analyzed in our study), may represent the earliest divergent representatives of the genus Rosa. Their close relationships were reconstructed in two other phylogenetic studies of the genus (Wissemann and Ritz, 2005; Fougère-Danezan et al., 2015). We suppose they can be the only remaining descendants of an ancient group of roses, which is nearly completely extinct now, but was widely distributed in the Northern Hemisphere in the Oligocene (Becker, 1963; Kvaček and Walther, 2004; Kellner et al., 2012). Though these species are not morphologically similar and formerly were placed into two different subgenera, Hulthemia and Hesperhodos, respectively, they do share some similar traits. All three species possess hypanthia with unusually wide orifices (up to 4−5 mm) externally covered with long acute bristles. Species from other clades usually have much more enclosed hypanthia with orifices not exceeding 2−3 mm in diameter, with the exception of R. roxburghii discussed below.
The haplotype 6 (R. spinosissima) is basal for two haplogroups or lineages III and IV. Lineage III was represented in our study exclusively by haplotypes of R. kokanica, a yellow-flowered Central Asian species. The unusual haplotypic diversity of this species represented by 12 haplotypes is worth special study. We suppose it could have originated from a long period of isolation of populations of this species in the foothills of the Tien Shan mountains during Pleistocene glaciations. The lineage or haplogroup IV is represented by seven haplotypes of species belonging to the sections Rosa, Synstylae, and Caninae distributed in different parts of the wide range of the genus, and numerous intermediate haplotypes absent in our sample. We suppose they may represent remnants of some ancient and mostly extinct haplotype group that partly survived because of hybridization.
Thus, the haplotype 6 appears to be basal for haplogroups III and IV, being the root haplotype for the haplogroup II and differing from its central and the most widely distributed haplotype 26 by a single mutation. The latter is the central interior haplotype of the haplogroup II and the whole network as well. Actually, it is the root haplotype for the two main lineages of Rosa represented, on the one hand, by the haplogroup V uniting the majority of members of the section Rosa, and on the other hand, by the remaining haplogroups VI to IX, uniting members of the sections Microphyllae, Banksianae, Laevigatae, Synstylae, Gallicanae, and Caninae.
Sequences of accessions of the section Pimpinellifoliae occur only in haplogroups II and III, while the bulk of sequences of the Rosa section members belong to haplogroups VI and V. Nevertheless, haplotypes of several species of the section Rosa, such as R. prattii, R. moyesii, R. acicularis, and R. macrophylla, belong to haplogroup II. This fact may be explained by incomplete lineage sorting, as in the case of R. macrophylla (interior haplotype 26), or by hybridization or wrong sectional taxonomic identification as in the case of the other three species (tip haplotypes 31, 35, 37). In particular, R. acicularis is represented in our network by unrelated or weakly related haplotypes 12, 13, and 31, which can be explained by hybridization and extinction of the majority of haplotypes in this circumboreal highly polyploid species in glacial times. The species deserves special study using adequately larger material. Rosa praelucens (section Microphyllae), another high polyploid (decaploid: 2n = 10x = 70; Jian et al., 2010) species endemic to Yunnan Province of China, possesses interior haplotype 26, which most probably may be due to multiple hybridization events with species of the sections Rosa and/or Pimpinellifoliae (Fougère-Danezan et al., 2015). This species is very similar to the diploid R. roxburghii in its morphological traits; however, in phylogenetic trees reconstructed using plastid markers, it repeatedly appears in a clade shaped by species of the section Rosa (Fougère-Danezan et al., 2015; Cui et al., 2022). Species of the section Synstylae, possessing tip haplotypes 8, 10, and 28, most likely appear in haplogroups II and IV also due to hybridization events with members of sections Pimpinellifoliae and Rosa. We would like to mention specially the unstable position of the haplotype 51 represented by R. omeiensis in the TCS network and the NeighborNet split graph. That was the only sequence of the Chinese members of the section Pimpinellifoliae, to which R. sericea and R. xanthina also belong, in our study. This group often appears to be the earliest divergent in full plastome studies. This fact indicates the heterogeneity of the section Pimpinellifoliae, which probably includes various descendants of the initial group of roses morphologically similar due to symplesiomorphies.
Haplogroups VII to IX unite all accessions from the sections Synstylae, Chinenses, Gallicanae, and Caninae. These haplogroups are evidently derived from the haplogroup II which bears the central position in our haplotype network. The haplogroup VI is positioned between them and evidently includes the remnants of a now nearly extinct group of roses, intermediate between the sections Pimpinellifoliae and Synstylae. Due to numerous missing intermediate haplotypes present in this group, the relationships between them are less certain. However, according to this result, R. roxburghii (section Microphyllae, haplotype 47) seems to be ancestral to species of sections Banksianae and Laevigatae. These species also possess widely open hypanthia with a large orifice, a character rather primitive from the morphological point of view.
The haplogroup VII encompasses the majority of species of the section Synstylae and all species of the section Chinenses. The latter chiefly differs from the former by free styles; in species of the section Synstylae, they are connate in a column. Though being free in members of the section Chinenses, they are still long protruding from the narrow hypanthium orifice, thus strongly resembling members of Synstylae in this feature. All accessions of R. chinensis share interior haplotype 58 with the majority of Synstylae accessions; R. odorata possesses tip haplotypes 56 and 57, and R. lucidissima possesses tip haplotypes 61 and 63, all derived from haplotype 58. Thus, the section Chinenses is not monophyletic and, probably, should be lumped with the section Synstylae.
Haplotypes of the section Caninae clearly shape two different lineages (haplogroups VIII and IX) derived from haplotype 58, i.e., from the section Synstylae. In both cases, basal interior haplotypes 64 and 71 are shared with the West Eurasian members of Synstylae. In the case of haplogroup VIII, this is R. sempervirens distributed in the Mediterranean. In the case of haplogroup IX, they are the European R. arvensis and the West Asian R. phoenicia, sharing the haplotype 71 also with R. gallica of the section Gallicanae. This finding corroborates previous results by other authors (Fougère-Danezan et al., 2015; Zhu et al., 2015), who also had West Asian and European species of Synstylae that appeared in the same clades as the species of the Caninae section. The fact that western species of Synstylae possess basal haplotypes of the two branches of the Caninae section makes them or their immediate ancestors plausible ancestral species for this section. Thus, it appears that the so-called ‘Canina-meiosis’ has appeared at least twice independently. This idea is supported also by detailed cytogenetic studies of several West European species of the section Caninae (Herklotz et al., 2018; Lunerová et al., 2020).
Peculiar Properties of Evolution of the Genus Rosa
Our analysis suggests broadly paraphyletic relationships among major taxonomic groups of roses, the Synstylae–Caninae group (haplogroups VII to IX) and Rosa (haplogroups III and V) being a derivative of the Pimpinellifoliae group (II and IV).
These conclusions are supported by the geographic distributions of haplogroups and taxonomic sections (Fig. 2). The haplogroup I and species of the section Minutifoliae have a disjunctive distribution in Central Asia and western North America, correspondingly. We argue such a range of the group confirms its relic nature.
Rosa fossils are found in sediments starting from the upper Eocene, in the Oligocene roses are known from numerous localities in Asia, Europe, and North America (Becker, 1963; Kvaček and Walther, 2004; Kellner et al., 2012). These fossils are, however, represented mostly by separate leaflets of compound leaves, spines, and very rarely flowers, fruits, or intact compound leaves, which enables their taxonomic identification to the genus level only. Molecular dating of a phylogenetic tree (Fougère-Danezan et al., 2015) does not contradict these data. According to it, diversification of the main clades occurred in the middle to upper Oligocene. Divergence of the clade R. minutifolia–R. persica occurred approximately at the same time. Diversification of modern sections occurred from Miocene to Pliocene. Thus, the Oligocene age fossils are incomparable with modern taxonomic sections, and the early divergent R. minutifolia and R. persica may represent descendants of the initial extinct group of roses. Haplogroups II to V, representing taxonomic sections Pimpinellifoliae and Rosa, are widely distributed in Eurasia and North America. Though species of the former section are absent from North America, in Eurasia their range is wide and discontinuous. In our minds, such a range marks the long persistence of the group throughout its range. Contrary to that, the geographic distribution of haplogroups VII to IX representing taxonomic sections Synstylae, Chinenses, Gallicanae, and Caninae is continuous (with an exception for the North American R. setigera) and more restricted. The range of the section Synstylae is mostly restricted to southern and eastern China with a few species reaching West Asia and Europe. The range of the section Caninae is restricted to Europe and West and Central Asia. The oligotypic sections Chinenses and Gallicanae have smaller ranges included within major ranges of their corresponding haplogroups. Such a type of the ranges indicates the relatively recent origin of the above mentioned sections, not earlier than the Pliocene, i.e., five to six million years, according to the results of molecular dating (Fougère-Danezan et al., 2015).
Thus, the results of our study lead us to the conclusion that the taxonomic system of the genus Rosa as such does not need reconsideration, since it adequately reflects the existing groups of related species. However, the taxonomic position of separate species should be reconsidered. The less taxonomically and phylogenetically studied section Pimpinellifoliae is, to our minds, ancestral to all the other sections of the genus, except for the descendants of the most ancient and nearly extinct group represented in modern times by two North American (section Minutifoliae) and one Central Asian (R. persica) species. Paraphyly of the majority of the groups is due to their recent origin and rapid diversification coupled with active hybridization between young species, which lead to the modern co-occurrence of ancestral groups and their descendants.
REFERENCES
Becker, H.F., The fossil record of the genus Rosa, Bull. Torrey Bot. Club, 1963, vol. 90, pp. 99–110.
Borchsenius, F., FastGap 1.2, Department of Biosciences, Aarhus University, Denmark, 2009. http://www.aubot.dk/FastGap_home.htm.
Bruneau, A., Starr, J.R., and Joly, S., Phylogenetic relationships in the genus Rosa: new evidence from plastid DNA sequences and an appraisal of current knowledge, Syst. Bot., 2007, vol. 32, no. 2, pp. 366–378. https://doi.org/10.1600/036364407781179653
Buzunova, I.O., Rosa L., in Flora Vostochnoi Evropy (Flora of Eastern Europe), Tsvelev, N.N., Ed., St. Petersburg: Mir i Sem’ya, 2001, vol. 10, pp. 329–361.
Chen, X., Liu, Y., Sun, J., Wang, L., and Zhou, S., The complete plastid genome sequence of Rosa acicularis in Rosaceae, Mitochondrial DNA, Part B, 2019a, vol. 4, no. 1, pp. 1743–1744. https://doi.org/10.1080/23802359.2019.1610100
Chen, M., Zhang, C., and Gao, X., The complete plastid genome sequence of Rosa pricei (Rosaceae), Mitochondrial DNA, Part B, 2019b, vol. 4, no. 1, p. 1918.
Clement, M., Posada, D., and Crandall, K.A., TCS: a computer program to estimate gene genealogies, Mol. Ecol., 2000, vol. 9, pp. 1657–1659. https://doi.org/10.1046/j.1365-294x.2000.01020.x
Crandall, K.A. and Templeton, A.R., Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction, Genetics, 1993, vol. 134, pp. 959–969. https://doi.org/10.1093/genetics/134.3.959
Crépin, F., Primitiae monographiae Rosarum. Matériaux pour servir a l’histoire des Roses, Gand, Marché aux Grains: Annoot-Braeckman, 1869.
Crépin, F., Nouvelle classification des roses, Extrait du Journal des Roses, Drosne: Melun E., 1891, vols. 3–5, pp. 3–30.
Criscuolo, A. and Gribaldo, S., BMGE (block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments, BMC Evol. Biol., 2010, vol. 10, p. 210. https://doi.org/10.1186/1471-2148-10-210
Cui, W.H., Zhong, M.C., Du, X.Y., Qu, X.J., Jiang, X.D., Sun, Y.B., Wang, D., Chen, S.Y., and Hu, J.Y., The complete plastid genome sequence of a rambler rose, Rosa wichuraiana (Rosaceae), Mitochondrial DNA, Part B, 2020, vol. 5, no. 1, p. 252. https://doi.org/10.1080/23802359.2019.1700198
Cui, W.H., Du, X.Y., Zhong, M.C., Fang, W., Suo, Z.Q., Wang, D., Dong, X., Jiang, X.D., and Hu, J.Y., Complex and reticulate origin of edible roses (Rosa, Rosaceae) in China, Hortic. Res., 2022, vol. 9, p. uhab051. https://doi.org/10.1093/hr/uhab051
Debray, K., Marie-Magdelaine, J., Ruttink, T., Clotault, J., Foucher, F., and Malecot, V., Identification and assessment of variable single-copy orthologous (SCO) nuclear loci for low-level phylogenomics: a case study in the genus Rosa (Rosaceae), BMC Evol. Biol, 2019, vol. 19, p. 152. https://doi.org/10.1186/s12862-019-1479-z
Edler, D., Klein, J., Antonelli, A., and Silvestro, D., raxmlGUI 2.0 beta: a graphical interface and toolkit for phylogenetic analyses using RAxML, bioRxiv, 2019. https://doi.org/10.1101/800912
Eriksson, T., Hibbs, M.S., Yoder, A.D., Delwiche, C.F., and Donoghue, M.J., The phylogeny of Rosoideae (Rosaceae) based on sequences of the internal transcribed spacers (ITS) of nuclear ribosomal DNA and the trnL/F region of plastid DNA, Int. J. Pl. Sci., 2003, vol. 164, no. 2, pp. 197–211. https://doi.org/10.1086/346163
Fedorova, A.V., Schanzer, I.A., and Kagalo, A.A., Local differentiation and hybridization in populations of wild roses in W Ukraine, Wulfenia, 2010, vol. 17, pp. 99–115.
Fougère-Danezan, M., Joly, S., Bruneau, A., Gao, X.F., and Zhang, L.B., Phylogeny and biogeography of wild roses with specific attention to polyploids, Ann. Bot. (Oxford), 2015, vol. 115, pp. 275−291. https://doi.org/10.1093/aob/mcu245
Gurushidze, M., Fritsch, R.M., and Blattner, F.R., Species-level phylogeny of Allium subgenus Melanocrommyum: incomplete lineage sorting, hybridization and trnF gene duplication, Taxon, 2010, vol. 59, no. 3, pp. 829–840. https://doi.org/10.2307/25677671
Hall, T.A., BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT, Nucleic Acids Symp. Ser., 1999, vol. 41, pp. 95–98.
Henker, H., 25. Rosa, in Gustav Hegi—Illustrierte Flora von Mitteleuropa, Weber, H.E., Ed., Berlin: Parey Buchverlag, 2003, 2nd ed., vol. 4, no. 2C, pp. 1–108.
Herklotz, V., Kovařík, A., Lunerová, J., Lippitsch, S., Groth, M., and Ritz, C.M., The fate of ribosomal RNA genes in spontaneous polyploid dogrose hybrids [Rosa L. sect. Caninae (DC.) Ser.] exhibiting non-symmetrical meiosis, Plant J., 2018, vol. 94, pp. 77–90. https://doi.org/10.1111/tpj.13843
Hudson, R.R., Gene genealogies and the coalescent process, Oxford Surv. Evol. Biol., 1990, vol. 7, pp. 1–44.
Huson, D.H., SplitsTree: a program for analyzing and visualizing evolutionary data, Bioinformatics, 1998, vol. 14, pp. 68–73.
Huson, D.H. and Bryant, D., Application of phylogenetic networks in evolutionary studies, Mol. Biol. Evol., 2006, vol. 23, pp. 254–267.
International Code of Botanical Nomenclature (Vienna Code), McNeil, J., Barrie, C.F.R., Burdet, H.M., et al., Eds., Ruggell, Liechtenstein: Gantner Verlag, 2006.
International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen Code) Adopted by the Nineteenth International Botanical Congress, Shenzhen, China, July 2017, Turland, N.J., Wiersema, J.H., Barrie, F.R., Greuter, W., Hawksworth, D.L., Herendeen, P.S., Knapp, S., Kusber, W.-H., Li, D.-Z., Marhold, K., May, T.W., McNeill, J., Monro, A.M., Prado, J., Price, M.J., and Smith, G.F., Eds., Regnum Veg., Glashütten: Koeltz Botanical Books, 2018, vol. 159. https://doi.org/10.12705/Code.2018
Jacob, S.S. and Blattner, F.R., A plastid genealogy of Hordeum (Poaceae): long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference, Mol. Biol. Evol., 2006, vol. 23, no. 8, pp. 1602–1612. https://doi.org/10.1093/molbev/msl018
Jeon, J.-H. and Kim, S.C., Comparative analysis of the complete chloroplast genome sequences of three closely related East-Asian wild roses (Rosa sect. Synstylae, Rosaceae), Genes (Basel), 2019, vol. 10, no. 1, p. 23. https://doi.org/10.3390/genes10010023
Jian, H.-Y., Zhang, H., Tang, K.-X., Li, S.-F., Wang, Q.-G., Zhang, T., Qiu, X.-Q., and Yan, H.-J., Decaploidy in Rosa praelucens Byhouwer (Rosaceae) endemic to Zhongdian Plateau, Yunnan, China, Caryologia, 2010, vol. 63, pp. 162–167.
Jian, H.Y., Zhang, Y.H., Yan, H.J., Qiu, X.Q., Wang, Q.G., Li, S.B., and Zhang, S.D., The complete plastid genome of a key ancestor of modern roses, Rosa chinensis var. spontanea, and a comparison with congeneric species, Molecules, 2018a, vol. 23, p. 389. https://doi.org/10.3390/molecules23020389
Jian, H.Y., Zhang, S., Zhang, T., Qui, X.Q., Yan, H.J., Li, S.B., Wang, Q.G., and Tang, K.X., Characterization of the complete plastid genome of a critically endangered decaploid rose species, Rosa praelucens (Rosaceae), Conserv. Genet. Res., 2018b, vol. 10, pp. 851–854. https://doi.org/10.1007/s12686-017-0946-3
Katoh, K. and Standley, D.M., MAFFT multiple sequence alignment software version 7: improvements in performance and usability, Mol. Biol. Evol., 2013, vol. 30, pp. 772–780. https://doi.org/10.1093/molbev/mst010
Katoh, K., Misawa, K., Kuma, K., and Miyata, T., MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform, Nucleic Acids Res., 2002, vol. 30, pp. 3059–3066. https://doi.org/10.1093/nar/gkf436
Kellner, A., Benner, M., Walther, H., Kunzmann, L., Wissemann, V., and Ritz, C.M., Leaf architecture of extant species of Rosa L. and the Paleogene species Rosa lignitum Heer (Rosaceae), Int. J. Plant Sci., 2012, vol. 173, no. 3, pp. 239–250.
Kinene, T., Wainaina, J., Maina, S., and Boykin, L.M., Rooting trees, methods for, in Encyclopedia of Evolutionary Biology, Kliman, R.M., Ed., Oxford: Academic, 2016, vol. 3, pp. 489−493.
Klášterský, I., Rosa L., in Flora Europaea, Tutin, T. et al., Eds., Cambridge: Cambridge Univ. Press, 1968, vol. 2, pp. 25–32.
Koopman, W.J.M., Wissemann, V., De Cock, K., Van Huylenbroeck, J., De Riek, J., Sabatino, G.J.H., Visser, D., Vosman, B., Ritz, C.M., Maes, B., Werlemark, G., Nybom, H., Debener, T., Linde, M., and Smulders, M.J.M., AFLP markers as a tool to reconstruct complex relationships: a case study in Rosa (Rosaceae), Am. J. Bot., 2008, vol. 95, no. 3, pp. 353–366. https://doi.org/10.3732/ajb.95.3.353
Ku, T.C. and Robertson, K.R., Rosa Linnaeus, in Flora of China, Wu, C.Y. and Raven, P.H., Eds., Beijing/St. Louis: Science Press/Missouri Botanical Garden Press, 2003, vol. 9, pp. 339–381.
Kvaček, Z. and Walther, H., Oligocene flora of Bechlejovice at Děčín from the neovolcanic area of the České středohoří Mountains, Czech Republic, Acta Mus. Nat. Pragae, Ser. B, Hist. Nat., 2004, vol. 60, nos. 1–2, pp. 9–60.
Lewis, W.H., Ertter, B., and Bruneau, A., Rosa L., Flora of North America north of Mexico, Editorial Committee, Eds., vol. 9. http://www.efloras.org/florataxon.aspx?flora_id =1&taxon_id=128746. Accessed November 10, 2022.
Linnaeus, C., Species Plantarum, Holmiae: Impensis Laurentii Salvii, 1753, vol. 1, pp. 491–492.
Liu, C., Wang, G., Wang, H., Xia, T., Zhang, S., Wang, Q., and Fang, Y., Phylogenetic relationships in the genus Rosa revisited based on rpl16, trnL-F, and atpB-rbcL sequences, Hort. Sci., 2015, vol. 50, no. 11, pp. 1618–1624.
Lunerová, J., Herklotz, V., Laudien, M., Vozárová, R., Groth, M., Kovařík, A., and Ritz, C.M., Asymmetrical canina meiosis is accompanied by the expansion of a pericentromeric satellite in non-recombining univalent chromosomes in the genus Rosa, Ann. Bot. (Oxford), 2020, vol. 125, no. 7, pp. 1025–1038. https://doi.org/10.1093/aob/mcaa028
Meng, J., Fougere-Danezan, M., Zhang, L.-B., Li, D.-Z., and Yi, T.-S., Untangling the hybrid origin of the Chinese tea-roses: evidence from DNA sequences of single-copy nuclear and chloroplast genes, Plant Syst. Evol., 2011, vol. 297, pp. 157–170. https://doi.org/10.1007/s00606-011-0504-5
Meng, J., Jiang, H., Linna Zhang, L., and He, J., Characterization of the complete plastid genome of an important Chinese Old Rose Rosa odorata var. pseudindica, Mitochondrial DNA, 2019, vol. 4, no. 1, pp. 679–680. https://doi.org/10.1080/23802359.2019.1572469
Peterson, A., Harpke, D., Peterson, J., Harpke, A., and Peruzzi, L., A pre-Miocene Irano-Turanian cradle: origin and diversification of the species-rich monocot genus Gagea (Liliaceae), Ecol. Evol., 2019, vol. 9, no. 10, pp. 5870–5890. https://doi.org/10.1002/ece3.5170
Plants of the World online (POWO). http://plantsoftheworldonline.org/. Accessed August 7, 2022.
Potter, D., Gao, F., Bortiri, P.E., Oh, S.-H., and Baggett, S., Phylogenetic relationships in Rosaceae inferred from chloroplast matK and trnL-trnF nucleotide sequence data, Plant Syst. Evol., 2002, vol. 231, pp. 77–89. https://doi.org/10.1007/s006060200012
Rehder, A., Rosa L., Manual of Cultivated Trees and Shrubs Hardy in North America, New York: Macmillan, 1949, 2nd ed., pp. 426–451.
Ritz, C.M., Schmuths, H., and Wissemann, V.. Evolution by reticulation: European dogroses originated by multiple hybridization across the genus Rosa, J. Hered., 2005, vol. 96, no. 1, pp. 4–14.
Schanzer, I.A., Phylogeny and taxonomy of recently diverged groups as exemplified by the genus Rosa, Tr. Zool. Inst. Ross. Akad. Nauk, 2011, Appendix 2, pp. 202–216.
Schanzer, I.A., Vagina, A.V., and Ostapko, V.M., A critical study of wild roses (Rosa L.) from the “Khomutovskaya Steppe” Reserve, Byull. Mosk. O-va Ispyt. Prir., Otd. Biol., 2011, vol. 116, no. 3, pp. 38–49.
Schanzer, I.A., Fedorova, A.V., Galkina, M.A., Chubar, E.A., Rodionov, A.V., and Kotseruba, V.V., Is Rosa × archipelagica (Rosaceae, Rosoideae) really a spontaneous intersectional hybrid between R. rugosa and R. maximowicziana? Molecular data confirmation and evidence of paternal leakage, Phytotaxa, 2020, vol. 428, no. 2, pp. 93–103. https://doi.org/10.11646/phytotaxa.428.2.3
Shaw, J., Lickey, E.B., Schilling, E.E., and Small, R.L., Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III, Am. J. Bot., 2007, vol. 94, no. 3, pp. 275–288. https://doi.org/10.3732/ajb.94.3.275
Silvestro, D. and Michalak, I., raxmlGUI: a graphical front-end for RAxML, Organisms Diversity Evol., 2012, vol. 12, pp. 335–337. https://doi.org/10.1007/s13127-011-0056-0
Simmons, M.P. and Ochoterena, H., Gaps as characters in sequence-based phylogenetic analyses, Syst. Biol., 2000, vol. 49, no. 2, pp. 369–381. https://doi.org/10.1093/sysbio/49.2.369
Templeton, A.R., Crandall, K.A., and Sing, C.F., A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation, Genetics, 1992, vol. 132, pp. 619–633.
The World Checklist of Vascular Plants (WCVP), Govaerts, R.H.A., Ed. https://wcvp.science.kew.org/. Accessed August 7, 2022.
Tomljenović, N. and Pejić, I., Taxonomic review of the genus Rosa, Agric. Conspect. Sci., 2018, vol. 83, no. 2, pp. 139–147. https://hrcak.srce.hr/203011
Wang, Q., Hu, H., An, J., Bai, G., Qunli Ren, Q., and Liu, J., Complete plastid genome sequence of Rosa roxburghii and its phylogenetic analysis, Mitochondrial DNA, Part B, 2018, vol. 3, no. 1, pp. 149–150. https://doi.org/10.1080/23802359.2018.1431074
Wang, M., Zhang, C., Li, M., and Gao, X., The complete plastid genome sequence of Rosa banksiae var. normalis (Rosaceae), Mitochondrial DNA, Part B, 2019, vol. 4, no. 1, pp. 969–970. https://doi.org/10.1080/23802359.2019.1580163
Wissemann, V., Conventional taxonomy (wild roses), in Encyclopedia of Rose Science, Roberts, A.V., Debener, T., and Gudin, S., Eds., Amsterdam: Elsevier, 2003, pp. 111–117.
Wissemann, V., Beauty and the bastards. Intensive hybridization controls the evolution of wild roses, B.I.F. FUTURA, 2006, vol. 21, pp. 158–163.
Wissemann, V. and Ritz, C., The genus Rosa (Rosoideae, Rosaceae) revisited: molecular analysis of nrITS-1 and atpB-rbcL intergenic spacer (IGS) versus conventional taxonomy, Bot. J. Linn. Soc., 2005, vol. 147, pp. 275–290. https://doi.org/10.1111/j.1095-8339.2005.00368.x
Yin, X., Liao, B., Guo, S., Liang, C., Pei, J., Xu, J., and Chen, S., The plastids genomic analyses of Rosa laevigata, R. rugosa and R. canina, Chin. Med., 2020, vol. 15, p. 18. https://doi.org/10.1186/s13020-020-0298-x
Zhang, J., Esselink, G.D., Che, D., Fougère-Danezan, M., Arens, P., and Smulders, M.J.M., The diploid origins of allopolyploid rose species studied using single nucleotide polymorphism haplotypes flanking a microsatellite repeat, J. Hort. Sci. Biotechnol., 2013, vol. 88, no. 1, pp. 85–92. https://doi.org/10.1080/14620316.2013.11512940
Zhang, S.D., Jin, J.J., Chen, S.Y., Chase, M.W., Soltis, D.E., Li, H.T., Yang, J.B., Li, D.Z., and Yi, T.S., Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics, New Phytol., 2017, vol. 214, no. 3, pp. 1355–1367. https://doi.org/10.1111/nph.14461
Zhang, C., Xiong, X., and Gao, X., The complete plastid genome sequence of Rosa laevigata (Rosaceae), Mitochondrial DNA, Part B, 2019, vol. 4, no. 2, pp. 3556–3557. https://doi.org/10.1080/23802359.2019.1674200
Zhang, S.D., Zhang, C., and Ling, L.Z., The complete plastid genome of Rosa berberifolia, Mitochondrial DNA, 2019, vol. 4, no. 1, pp. 1741–1742. https://doi.org/10.1080/23802359.2019.1610093
Zhang, C., Li, S.-Q., Xie, H.-H., Liu, J.-Q., and Gao, X.-F., Comparative plastid genome analyses of Rosa: insights into the phylogeny and gene divergence, Tree Genet. Genomes, 2022, vol. 18, p. 20. https://doi.org/10.1007/s11295-022-01549-8
Zhao, X. and Gao, C., The complete plastid genome of Rosa minutifolia, Mitochondrial DNA, Part B, 2020, vol. 5, no. 3, pp. 3320–3321. https://doi.org/10.1080/23802359.2020.1817807
Zhao, L., Zhang, H., Wang, Q.-G., Ma, C.L., and Jian, H.-Y., The complete plastid genome of Rosa lucidissima, a critically endangered wild rose endemic to China, Mitochondrial DNA, Part B, 2019, vol. 4, no. 1, pp. 1826–1827. https://doi.org/10.1080/23802359.2019.1613198
Zhu, Z.M., Gao, X.F., and Fougère-Danezan, M., Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on plastid and nuclear markers, Mol. Phylogenet. Evol., 2015, vol. 87, pp. 50–64. https://doi.org/10.1016/j.ympev.2015.03.014
ACKNOWLEDGMENTS
The authors are grateful to the following colleagues who helped with collecting samples in the wild from various locations: K. Tojibaev (Botanical Institute, Uzbekistan), G. Šramko (Debrezen University, Hungary), E.A. Chubar (Natural Science Center for Marine Biology, Russia), Y.K. Vinogradova (Tsitsin Main Biological Garden, Russia), V. Herklotz (Senckenberg Natural History Museum, Germany), V.M. Ostapko (Donetzk Biological Garden, DPR), A.A. Kagalo (Institute of Ecology of the Carpathians, Ukraine), I. Bartish (Institute of Botany, Czech Republic), M.G. Pimenov (Moscow State University, Russia), A. Elkordy (Sohag University, Egypt), and curators of the Herbaria MHA, LE, KW, B, BP, and DE for permissions to destructive sampling of specimens. We thank K. Chamberlain (United Kingdom) for his useful linguistic corrections of the text and I.V. Belyaeva (Kew Gardens, United Kingdom) for her comments and suggestions for the manuscript.
Funding
This study was supported by the Tsitsin Main Botanical Garden, State Assignment no. 122042700002-6, and grants nos. 16-04-01390 and 19-04-01308 from the Russian Foundation for Basic Research. The authors are also grateful to the Ministry of Science and Higher Education of the Russian Federation for financial support of the Common Facilities Center “Herbarium of the Main Botanical Garden, Russian Academy of Sciences,” grant no. 075-15-2021-678.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This work does not contain any studies involving human and animal subjects.
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
Additional information
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Schanzer, I.A., Fedorova, A.V. & Meschersky, I.G. A Haplotype Network Approach to Reconstruct the Phylogeny of Rosa L. (Rosaceae). Biol Bull Russ Acad Sci 51, 331–345 (2024). https://doi.org/10.1134/S106235902360469X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S106235902360469X