
Abstract—
Preconcentration of strontium and barium as complexes with 11 organic reagents by coprecipitation with organic co-precipitants for their subsequent determination by X-ray fluorescence spectrometry is studied. The most effective systems turned out to be those included reagents from bisazosubstituted chromotropic acids, nitchromazo and chlorophosphonazo III. Complexes of these metals are virtually quantitatively coprecipitated as pairs with cations of the brilliant green dye, of the collector is an associate of an excess of the analytical reagent with cations of this dye. It is shown that the additional use of polyvinyl butyral as an indifferent co-precipitant ensures not only the almost complete extraction of these elements from solutions, but also the preparation of emitter concentrates suitable for X-ray fluorescence measurements using the standard-background technique. High efficiency allows, under optimal conditions, the achievement of very low limits of detection (IUPAC): 0.03 μg/mL Sr and 0.19 μg/mL Ba, even in working with small samples (by volume).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The severe consequences that arise from the long-term intake of strontium and barium into a human body make it necessary to monitor the contents of these elements in environmental samples and introduce the latest analytical methods for studying various types of waters, such as sea and river ones [1–5]. Because of the low contents of elements in these samples [6, 7], it is necessary not only to use the most sensitive methods, but also to combine them with selective preconcentration. Additional motives may include data on biogeochemical provinces for strontium and barium [8] and the new “Manhattan Project” [9].
Well-known and sensitive methods for determining strontium and barium are flame emission photometry [10], inductively coupled plasma atomic emission spectrometry [1, 3, 4, 11–13], including preconcentration on a selective sorbent [14], X-ray fluorescence preconcentration analysis [15] and other newer methods, such as ion chromatography [16] and potentiometry with ion-selective electrodes [17]. The features and limitations of these methods for determining small amounts of elements in various waters are well known. Thus, in flame emission photometry this is the ionization of elements at low concentrations, anion interferences, including for aluminum, which forms difficult-to-dissociate aluminates in flames, which makes it difficult to use resonance lines [10], overlapping with cyan bands in the case of strontium coincide. Better effects can be achieved by combining more selective X-ray fluorescence (XRF) spectrometry with preconcentration procedures. Among the latter, the greatest attention is paid to sorption preconcentration, but the achieved effect is not always successful specifically for strontium and barium [15]. To solve other problems, a combination of XRF with preconcentration by coprecipitation attracts attention, for which, a gain in sensitivity at high selectivity may be more effective than in other methods [15]. However, inorganic coprecipitants for strontium turn out to be ineffective—strontium is unselectively coprecipitated with iron(III) hydroxide by only 20% [18], although standardized procedures for such coprecipitation from very large volumes are used in radiochemistry [19].
Significantly more effective and analytically diverse are options for coprecipitation with organic reagents, the principles and examples of which were outlined in the monograph [20], and their possibilities were discussed and evaluated in the monograph [21]. In this work, this method was extended to its combination with a significantly more selective XRF spectrometry. In preconcentration by coprecipitation, the transfer of small and ultra-small amounts of the elements being determined from the liquid phase into a compact solid phase becomes especially interesting. This guarantees high concentration coefficients, is well combined with the characteristics of XRF spectrometry in the preparation of emitter concentrates, and has not previously been used in combination with this preconcentration method. The novelty and efficiency of the proposed XRF preconcentration method, which allows the preconcentration of elements as their chelates using an indifferent co-precipitant, was supported by a patent [22]. A combination of the described techniques allows one to flexibly select the most suitable reaction with an organic analytical reagent for solving a practical problem, as well as an organic collector, and after the quantitative separation of the precipitate by filtration, prepare thin, mechanically strong, and compact films of emitter concentrates on filters, convenient for their analysis by XRF spectrometry. A possibility of using the preconcentration of strontium and barium by coprecipitation from individual solutions with some bisazosubstituted chromotropic acids was assessed by us earlier in [23].
The aim of this work was to study a combination of the highly efficient preconcentration of strontium and barium by the coprecipitation of their complexes with organic analytical reagents using organic co-precipitant collectors, which makes it possible to obtain compact emitter concentrates suitable for the sensitive and selective determination of small amounts of these elements by the X-ray fluorescence spectrometry.
EXPERIMENTAL
Solutions and reagents. As complexing reagents, we used organic analytical reagents (OR) with various functional analytical groups that give the most selective reactions with strontium and barium ions: reagents of the bisazosubstituted chromotropic acid group—arsenazo III, carboxyarsenazo, nitchromazo, chlorophosphonazo III [24]; xylenol orange; precipitating reagents represented by brilliant yellow and sodium rhodizonate [25]. The working scheme of coprecipitation according to the recommendations of [20] was chosen based on the nature of the complexes and analytical forms.
To precipitate excess reagents with sulfo groups as collectors in the “salt” mechanism of coprecipitation according to the data of [20], and ion associates of reagent complexes with strontium and barium ions, acridine orange, rhodamine 6G, and brilliant green dyes were used as precipitating cations. Polyvinyl butyral as an ethanol solution was an indifferent co-precipitant stimulating precipitation and co-precipitation [22]. Let us emphasize the cementing role of this component in the preparation of the emitter concentrate for XRF spectrometry during the filtering process. To ensure the aggregative and kinetic stability of the heterogeneous system, a 1% gelatin solution was added to a test solution before preconcentration; this ensured the uniform distribution of the concentrate over the filter surface.
The purity of all initial reagents and preparations in the solid form was controlled by XRF spectrometry, which made it possible to consider them as “pure for XRF.” When choosing the most efficient co-precipitant, we adhered to the requirements and algorithms formulated in the monograph [20], focusing on the specifics of the X-ray fluorescent ending. To avoid losses during preconcentration, OR forming the most stable complexes and possible masking were used. In performing concentration, a sufficiently low precipitation rate was taken into account, a possibility of the ripening of the concentrate precipitates and the optimization of the entire concentration procedure were taken into account. Constant pH values were maintained with buffer solutions: pH ≈ 5 with a 0.05 M acetate, pH ≈ 9 with a tartrate, and pH ≈ 12 with a 0.01 M NaOH solution. The pH values were monitored with a standard pH meter with a glass and a silver chloride electrode.
Equipment. XRF measurements were performed on an EDX3600 X-ray fluorescence energy dispersive spectrometer (Skyray, United States). Parameters of the spectrometer were as follows: voltage 40 kV, current 1000 mA, exposure time 40 s. We worked with the most sensitive spectral lines of K-series elements for strontium and L-series for barium [26]:
Element | Line | λ, nm | Quantum yield | Intensity, % |
|---|---|---|---|---|
Sr | Kα1 | 0.0875 | 0.702 | 100 |
Ba | Lα1 | 0.2776 | 0.093 | 100 |
Procedure for preparing emitter concentrates. A 10‑mL portion of a buffer solution with the required pH value were added to 30 mL of a test solution containing 0.3–2 μg/mL of the elements being determined. Next, in implementing the coprecipitation of element complexes with water-soluble reagents, 2 mL of a 0.1% solution of ORs, 3 mL of a 1% solution of gelatin, 2 mL of a 2% solution of a cationic dye, and 0.36 mL of a 15% ethanolic solution of polyvinyl butyral (PVB) were introduced in succession upon constant stirring. The resulting microheterogeneous system, after 15 min of exposure, was filtered under vacuum of a water-jet pump through a “blue ribbon” filter of a diameter of ~20 mm and pre-conditioned with a PVB sol in water [22], which was placed in a funnel with a Schott filter no. 2. Next, the filter with the concentrate was removed from the funnel, dried in an oven at a temperature of up to 100°C for 20 min and leveled in a book stocker. The prepared emitter concentrate was used for measurements by X-ray fluorescence spectrometry.
In using reagents forming compounds with metal ions poorly soluble in water (sodium rhodizonate, brilliant yellow), 10 mL of a buffer solution of a required pH value, a precipitant solution were added sequentially to 30 mL of a test solution containing 0.3–2 μg/mL of the test elements and then acted as described above.
RESULTS AND DISCUSSION
The use of OR of various classes under optimal conditions for performing analytical reactions required the use of appropriate coprecipitation schemes [20], adapted for the best quality of the prepared emitter concentrates.
Selection of a concentration scheme with organic reagents for subsequent analysis by X-ray fluorescence spectrometry. In the case of bisazosubstituted chromotropic acid and xylenol orange, to co-precipitate anionic complexes of strontium and barium, the formation of poorly water-soluble ion associates of these complexes with the dye cations was used. The collector in this case was an associate of a ~20-fold excess of the anionic reagent with an excess of the cationic dye. In the case of precipitating reagents of rhodizonate ions and brilliant yellow [25], co-precipitation was stimulated by introducing the indifferent PVB co-precipitant. Let us note that, because of the simultaneously cementing role of this component, its introduction turned out to be fundamentally important in all concentration options.
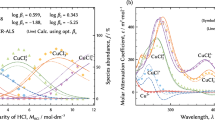
The efficiency of various OR during coprecipitation, taking into account the characteristics of the corresponding concentration systems, is compared in Table 1. One can see that, in the case of bisazosubstituted chromotropic acid, the efficiency of different suppliers of cations turned out to be different, which is confirmed by Fig. 1. The most successful coprecipitation systems turned out to be nitchromazo–brilliant green, pH ≈ 5, and chlorophosphonazo III–rhodamine 6G, pH ≈ 12. Apparently, this can be associated with the number of free anionic groups in OR complexes with strontium and barium ions and in the free reagents, available for the formation of poorly water-soluble associates. Moreover, at other things being equal, both systems are similar in their influence on the background at the corresponding energies of the strontium and barium lines, which is confirmed by Fig. 2. Based on the data [27], compared in Fig. 2, the background spectra can be associated with the manifestation of the dead time of the semiconductor detector of the energy-dispersive spectrometer due to the superposition of the read pulses (sum peaks). At the selected X-ray tube current, as can be seen in Fig. 1, this phenomenon weakly depends on the actual nature of the collector, although it is determined by the sample. The intensity of the scattered radiation is, of course, related to the number of light atoms, carbon and others, in the collector and in the indifferent co-precipitator.

X-ray fluorescence spectra of strontium and barium concentrates with some reagents. (a) Sr(II), Ba(II) (10 μg/mL, 5 mL)–nitchromazo (0.1%; 2 mL)–brilliant green cation (1%, 2 mL), pH ~ 5; (b) Sr2+, Ba2+ (10 μg/mL, 5 mL)–chlorophosphonazo(III) (0.1%, 2 mL)–rhodamine 6G cation (1%, 2 mL), pH ~ 12.

Comparison of background intensity in using the brilliant green cation and the rhodamine 6G cation. Complexing reagent chlorphosphonazo III, pH ~ 5, c(Sr2+) = 1.7 μg/mL, c(Ba2+) = 1.7 μg/mL, sample volume 30 mL. (1) brilliant green, (2) rhodamine 6G.
Nevertheless, the data in Table 2 and the achieved limits of detection allow us to draw clearer conclusions about the optimal systems for XRF spectrometry, which include the following components: nitchromzo−brilliant green, pH ≈ 5, chlorophosphonazo III−rhodamine 6G, pH ≈ 12. When choosing a preferred system in each specific case, one should take into account the chemical and analytical features of the OR. For example, in analyzing seawater samples, it is preferable to use the nitchromazo (better selectivity)−brilliant green, pH ≈ 5 system.
Co-precipitation chemistry. It is known that the process of co-precipitation occurs according to a complex multifactorial mechanism, especially with the participation of the indifferent co-precipitant PVB [15, 18, 20]. The formed complexes with the applied OR, depending on the type of the coprecipitation system under study, are entrapped by the precipitate with the resulting collector. In the case of bisazosubstituted chromotropic acid and xylenol orange derivatives, these are poorly water-soluble ion associates of excess reagents with cations of the main dyes (the “salt” mechanism according to the classification [20], which could more accurately be named the ion-associate mechanism). A favorable factor here can be considered the formal similarity of the structures of the associates of metal complexes and free reagent anions with the cations of the basic dyes. In the case of reagents forming poorly soluble neutral compounds with strontium and barium ions—brilliant yellow and rhodizonate ions—the use of promoting precipitants (indifferent according to the classification [19]) is required. Their efficiency is ensured by the fact of the formation of a fine PVB dispersion, which precipitates when a small amount of an ethanolic solution of this polymer is introduced into the sample at the final stage of coprecipitation. Let us also note that this ensures a certain aggregation and kinetic stability of the resulting heterogeneous system because of the addition of a gelatin solution to the sample as a stabilizer.
The quality of the emitter concentrate. The application of the proposed preconcentration procedure makes it possible to obtain emitter concentrates with an almost uniform distribution of the concentrated elements in a thin layer over the filter surface, as illustrated in Fig. 3. Conditioning the filter surface with a thin layer of PVB before filtration guarantees an almost complete separation of the concentrate precipitate from the mother liquor. The repeated control performance of all preconcentration procedures with the mother liquor showed the absence of lines of determined elements on the newly prepared “secondary” emitter concentrates. This indicates the almost complete extraction of elements by coprecipitation and is assessed as a criterion for the reliability of the studied preconcentration method, which guarantees high recovery of the elements and, accordingly, concentration coefficients.

Uniformity of the distribution of strontium over the surface of the concentrate. c(Sr2+) = 1.7 μg/mL, coprecipitation system nitchromazo–brilliant green, pH ~ 5. Sr, Kα (imp/s): (1) 10410, (2) 10391, (3) 10385, (4) 10403, (5) 10399.
Analytical features of various coprecipitation systems. From an analysis of calibration dependences in Table 2 and performance characteristics in Table 3 for various coprecipitation systems it follows that the best options are represented by OR and cationic dyes. In this case, the background is apparently due not only to the chemical composition of the thin-layer emitter on the filter, but also to the interaction of the primary radiation with the surface of the emitter concentrate, as well as the peculiarity of signal detection by the spectrometer [27]. The experimental conditions turned out to correspond well to the requirements of using the standard-background method, examples of calibration functions for which are given in Table 2: low concentrations of elements and almost constant composition of the matrix with respect to light elements [28]. One can also note the suitability of the technique for comparing different coprecipitation systems, because the established dependences can be attributed to a certain conditional “standard state of signals.” The given equations of calibration dependencies provide comparable estimates of relative standard deviations, correlation coefficients, and limits of detection for the elements.
Thus, the use of organic co-precipitants with good analytical selectivity ensures an almost complete isolation of elements from solutions even at their very low concentrations, which is known to be a feature of organic co-precipitants [20]. In this case, it becomes possible to extend the method to the determination of small amounts of elements in natural waters. We emphasize that the high selectivity of the determination of strontium and barium is ensured by a combination of the chemical component, OR in the coprecipitation systems and the X-ray fluorescence specificity of the signals, the optimal wavelengths and a possibility of using the thin layer approximation.
Examples of practical applications. The high concentration efficiency allows the developed method to be used for the determination of small amounts of strontium and barium in natural water samples. Table 4 shows an example of their preconcentration and determination in an artificial seawater sample without any pre-treatments other than those provided for in the procedure. As an experimental sample, we used a solution containing 35‰ sea salt, which corresponded to the approximate value of the salinity of sea water. If there is a strong influence of foreign elements, one can use masking with nitrilotriacetic acid in combination with the use of a weakly acidic medium and an appropriate preconcentration system. Let us note that most elements contained in natural waters do not interfere with the determination of strontium and barium in emitter concentrates using XRF spectrometry because of the absence of overlapping characteristic lines. For example, calcium, the content of which of natural waters is significantly higher than that of strontium and barium, has the most intense lines Kα 3.684 keV and Kβ 4.019 keV, while barium is determined by the most intense line Lα 4.472 keV and strontium, by the line Kα 14.143 keV.
To avoid the suppression of the coprecipitation of target ions, all reagents should be taken in large excesses, which allows small amounts of strontium and barium to be preconcentrated even in the presence of other elements in much higher concentrations. Large amounts of sulfate ions can have a certain effect on the coprecipitation of strontium and barium. However, the unstable outer-sphere associates they form with aqua ions of barium and especially strontium have little effect on the preconcentration of these elements. If necessary, using a column with an AB-17-8 ion exchanger in the Cl form at the sample preparation stage solves this problem.
We emphasize that the advantages of the discussed preconcentration technique in combination with XRF spectrometry make it possible to use very small sample volumes. Thus, e.g., in determining elements in extracts from bottom silt sediments, only 5 mL of a sample is sufficient, as illustrated by the results presented in Table 4 and in Fig. 4. The results of determining strontium and barium in a model seawater sample do not contradict the data on the average contents of these elements in seawater, 8 µg/mL Sr and 0.02 µg/mL Ba [6].

Spectrum of a concentrate of an experimental sample, close in composition to sea water. Nitchromazo–brilliant green system, pH 5, total sample volume 100 mL.
REFERENCES
Crompton, Th., Analysis of Oceanic Waters and Sediments, Boca Raton: CRC, 2016. https://doi.org/10.1201/b19088-4
Baker, R.A., Adv. Chem., 1968, vol. 73, p. 296. https://doi.org/10.1021/ba-1968-0073.ch018
Tarun, K.D. and Sarin, M., Geostand. Geoanal. Res., 2007, vol. 26, no. 3, p. 301. https://doi.org/10.1111/j.1751-908X.2002.tb00636.x
Prasada Rao, T., Metilda, P., and Mary Gladis, J., Crit. Rev. Anal. Chem., 2005, vol. 35, no. 4, p. 247. https://doi.org/10.1080/10408340500431272
Burton, J.H. and Price, T.D., J. Archaeol. Sci., 1990, vol. 17, p. 547. https://doi.org/10.1016/0305-4403(90)90035-4
Ivanov, V.V., Ekologicheskaya geokhimiya elementov: Spravochnik (Environmental Geochemistry of Elements: Handbook), Burenkov, E.K., Ed., Moscow; Nedra, 1994.
Gaillardet, J., Viers, J., and Dupre, B., in Treatise on Geochemistry, Turekian, K.K. and Holland, H.D., Eds., Amsterdam: Elsevier, 2014, vol. 7, p. 195.
Vinogradov, A.P., Polnoe sobranie trudov (Complete Collection of Works), 18 vols., Kostitsyn, Yu.A., Ed., vol. 4: Geokhimiya redkikh i rasseyannykh elementov v pochvakh (Geochemistry of Rare and Trace Elements in Soils), Korobov, E.M., Ed., Moscow: Ross. Akad. Nauk, 2021.
Kirby, P.K., Aluminum, Barium and Strontium: The New Manhattan Project Chemtrail Sprays, 2015. https:// www.activistpost.com/2015/07/aluminum-barium-and-strontium-new.html. Accessed December 7, 2022.
Chow, T.J. and Thompson, T., Anal. Chem., 1955, vol. 27, no. 1, p. 18. https://doi.org/10.1021/ac60097a006
Poluektov, N.S., Mishchenko, V.T., Kononenko, L.I., and Bel’tyukova, S.V., Analiticheskaya khimiya strontsiya (Analytical Chemistry of Strontium), Moscow: Nauka, 1978.
Frumina, N.S., Goryunova, N.N., and Eremenko, S.N., Analiticheskaya khimiya bariya (Analytical Chemistry of Barium), Moscow: Nauka, 1977.
Vasil’eva, M.A. and Polyakova, E.V., J. Anal. Chem., 2022, vol. 77, p. 1526. https://doi.org/10.1134/S1061934822120152
Ying, R., Int. J. Anal. Chem., 2015, vol. 2015, p. 425084. https://doi.org/10.1155/2015/425084
Margui, E., Van Grieken, R., Fontas, C., Hidalgo, M., and Queralt, I., Appl. Spectrosc. Rev., 2010, vol. 45, no. 3, p. 179. https://doi.org/10.1080/05704920903584198
Nesterenko, E.P., Nesterenko, P.N., Paull, B., Melendez, M., and Corredor, J.E., Microchem. J., 2013, vol. 111, p. 8. https://doi.org/10.1016/j.microc.2012.09.003
Chandra, S., Sharma, K., and Kumar, A., J. Saudi Chem. Soc., 2014, vol. 18, p. 555. https://doi.org/10.1016/j.jscs.2011.11.002
Wilson, I.D., Encyclopedia of Separation Science, Amsterdam: Elsevier, 2000.
Mundschenk, H., Procedures Manual for Monitoring of Radioactive Substances in the Environment and of External Radiation, 1994.
Kuznetsov, V.I. and Akimova, T.G., Kontsentrirovanie aktinoidov soosazhdeniem s organicheskimi soosaditelyami (Preconcentration of Actinides by Coprecipitation with Organic Coprecipitants), Kuznetsov, V.I., Ed., Moscow: Atomizdat, 1968.
Zolotov, Yu.A. and Kuz’min, N.M., Kontsentrirovanie mikroelementov (Preconcentration of Trace Elements), Moscow: Nauka, 1988.
Kuznetsov, V.V., Shalimova, E.G., Agudin, P.S., and Bespalov, E.L., RF Patent 2623194, 2017.
Prokopenko, Yu.R. and Kuznetsov, V.V., Usp. Khim. Khim. Tekhnol., 2020, vol. 34, no. 7, p. 11.
Savvin, S.B., Organicheskie reagenty gruppy arsenazo III (Organic Reagents of the Arsenazo III Group), Moscow: Atomizdat, 1971.
Savvin, S.B., Dedkova, V.P., and Akimova, T.G., Organicheskie reagenty na Ba 2+ i \(SO_{4}^{{2 - }}\) (Organic Reagents for Ba2+ and \({\text{SO}}_{4}^{{2 - }}\)), Moscow: Nauka, 1971.
Blokhin, M.A. and Shveitser, I.G., Rentgenospektral’nyi spravochnik (X-Ray Spectral Reference Book), Moscow: Nauka, 1982.
Mazuritskii, M.I., Physical foundations and methods of X-ray spectral studies. http://x-ray.sfedu.ru/Book_X-Ray_Tools.pdf. Accessed May 8, 2021.
Bakhtiarov, A.V. and Savel’ev, S.K., J. Anal. Chem., 2020, vol. 75, no. 1, p. 18. https://doi.org/10.1134/S1061934820010037
Funding
This work was supported by the Mendeleev University of Chemical Technology of Russia. No additional grants were obtained to conduct or direct this specific study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors of this work declare that they have no conflicts of interest.
Additional information
Translated by V. Kudrinskaya
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kuznetsov, V.V., Prokopenko, Y.R. Preconcentration of Strontium and Barium by Coprecipitation with Organic Collectors and Their Determination by X-Ray Fluorescence. J Anal Chem 79, 569–577 (2024). https://doi.org/10.1134/S1061934824050071
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934824050071