
Abstract
In this research, a preconcentration procedure was developed for the determination of silver in environmental samples using high-resolution continuum source graphite furnace atomic absorption spectrometry (HR-CS GFAAS). During the preconcentration step, bentonite was used as a cheap solid sorbent in ultrasound-assisted dispersive micro solid-phase extraction. The experimental parameters including pH of the sample solution, bentonite amount, and ultrasonication time as well as the main parameters of HR-CS GFAAS were investigated. The limit of detection was 0.01 µg/L, and the achieved preconcentration factor was 34. The relative standard deviation was 5%. The accuracy of this method was validated by analyses of NIST SRM 2709 (San Joaquin soil), NIST SRM 2711 (Montana soil), and NIST SRM 1643e (trace elements in water) certified reference materials. The proposed method was successfully applied for the determination of silver in soil and water samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The content of silver in environmental samples has increased due to its extensive use in the industry, especially as conductors, switches, and contacts in electrical applications, the catalyst in chemical reactions and for soldering. Silver is also used in medicine, photographic industry, jewelry, for the production of mirrors, coins, tableware, and batteries [1]. Recently, production of Ag nanoparticles for nanotechnology, medicine, food storage, textile coatings, and envi-ronmental applications has significantly increased [2].
Silver concentrations in environmental samples vary greatly and depend on geological area and distance from the anthropogenic sources of this element. The common range of mean contents of Ag in soils is between 0.06 and 0.4 mg/kg, but soils from mineralized areas are usually enriched in Ag. In world ocean waters, Ag concentration has been estimated in the range from 0.024 to 40 ng/L. Due to accumulation capacities of plants, Ag concentrations in them may be highly elevated when they grow in Ag-contaminated soils. This element is also relatively easily accumulated by aquatic biota that plays a crucial role in its cycling in aquatic environments, especially in its transfer to the food chain [3].
Silver is considered toxic for humans. The toxicity of Ag depends not only on its total concentration but also on its speciation. Silver is a reactive element in the aquatic environment, however, only the free ionic form (Ag+) is highly toxic at low concentrations [4]. The most common species of Ag occurring in soils are inorganic forms (e.g., Ag+, Ag2+, and AgO+) [3].
Due to the very low concentration of silver in environmental samples and the matrix interferences, the direct determination of this element by graphite furnace atomic absorption spectrometry (GFAAS) can be difficult [5]. Preconcentration and isolation of the analyte from the matrix can be achieved using the extraction techniques. Nowadays, nanomaterials are often used as solid sorbents [6].
Bentonite has a typical layered silicate structure consisting of two silica tetrahedral sheets and a central octahedral one. The interlayer space is easily accessible to water and other polar liquids [7]. This clay is characterized by biocompatibility, high surface area, eminent cation exchange capacity, and relatively low cost [8–11].
Dispersive micro solid-phase extraction (DMSPE) using various nanomaterials as solid sorbents is becoming more popular due to its many advantages, e.g., immediate interaction between metal ions and the nanomaterial as well as shorter time required for sample preparation in comparison with classical solid-phase extraction [12, 13].
Techniques such as liquid-liquid extraction [14], cloud point extraction [15], and dispersive liquid-liquid microextraction [16, 17] have been used so far to preconcentrate and separate trace Ag amount. The literature review has also revealed several papers related to the preconcentration of Ag on solid sorbents using solid-phase extraction prior to the determination by graphite furnace atomic absorption spectrometry. Silver has been successfully preconcentrated on alumina modified with polyethylenimine [18], silica gel modified with 3-aminopropyltriethoxysilane [19], magnetic nanocomposite modified with thiourea [20], carbon [21], chelating sorbent SPHERON® Thiol 1000 [22], and immobilized diethyldithiocarbamate (DDTC) on surfactant-coated alumina [23].
The purpose of this project was to improve the analytical potential of graphite furnace atomic absorption spectrometry with slurry sampling prior to Ag determination in environmental samples after dispersive micro solid-phase extraction using bentonite as a solid sorbent.
EXPERIMENTAL
Instrumentation. An Analytik Jena ContrAA 700 high-resolution atomic absorption spectrometer equipped with a 300 W xenon short-arc lamp (Analytic Jena, Jena, Germany) as a continuum radiation source was used throughout these experiments. In the research, a graphite furnace was used for Ag atomization. After the measurement, data were transferred to the computer for processing. The operating parameters of the high-resolution continuum source (HR-CS) GFAAS instrument are summarized in Table 1.
A UniClever focused microwave sample preparation system (Plazmatronika, Wrocław, Poland) operating at 2450 MHz and 300 W maximum output was used for certified reference materials and soils digestion. The computer-controlled system with continuous temperature, pressure, and microwave power monitoring was equipped with a high-pressure TFM-PTFE vessel and a water cooling system. The vessel capacity was 110 mL, and the maximum pressure and maximum temperature were 100 atm and 300°C, respectively.
Bentonite was weighed using an M2P microanalytical balance (Sartorius, Gottingen, Germany) with a resolution of 1 μg (electronic weighing range up to 2 g). The pH values were measured with a pH-meter (pH 211 Microprocessor, Hanna Instruments, Kehl, Germany) supplied with a combined glass electrode. A Sonopuls HD 70 ultrasonic cell disruptor/homogenizer (70 W, 20 kHz, Bandelin, Germany) equipped with a 2-mm titanium microtip was used for dispersive extraction before HR-CS GFAAS detection. A centrifuge (model EBA 20, Hettich, Germany) was employed for phase separation after the extraction procedure.
Gases and reagents. Compressed high-purity argon obtained from Air Products (Warsaw, Poland) was used as an inert gas. Working standard solutions of Ag were prepared from a 1000 mg/L atomic absorption standard solution (Merck, Darmstadt, Germany). Pd(NO3)2 and Mg(NO3)2 modifier stock solutions (10.0 ± 0.2 g/L for each element) were obtained from Merck. The pH of the sample solutions was adjusted with 65% (v/v) HNO3 and 30% (v/v) NaOH of the highest quality (Suprapur, Merck, Darmstadt, Germany). The bentonite (average particle size ≤25 µm) supplied from Aldrich (Steinheim, Germany) was used as a solid sorbent in dispersive micro solid-phase extraction. All mineral acids (65% (v/v) HNO3 and 40% HF (v/v)) and 30% H2O2 (v/v) of the highest quality (Suprapur, Merck, Darmstadt, Germany) were used for samples digestion. Deionized and double distilled water (quartz apparatus, Bi18, Heraeus, Germany) was used throughout the research. The water resistivity was 18 MΩ cm.
Certified reference materials and environmental samples. The accuracy of the method described in this study was evaluated using three certified reference materials (NIST SRM 2709, NIST SRM 2711, and NIST SRM 1643e) supplied by the National Institute of Standards and Technology, Gaithersburg, USA.
In this study, soil and water samples were analyzed. Soil samples were collected from the outer surface (10–20 cm) after removing surface contamination. A plastic spatula was used for sample collection. Then, soil samples were dried thoroughly. To ensure homogeneity, it was necessary to grind the solid samples. This was achieved in an agate pestle and mortar by the manual grinding of solids. After that, the samples were sieved through a <2 mm sieve and digested. The water samples were stored at 4°C in polyethylene flasks for a maximum of 7 days. The samples were filtered before analysis using a Cameo syringe filter with a polytetrafluoroethylene (PTFE) membrane and a pore size of about 0.22 mm (GE Water & Process Technologies, USA).
Microwave-assisted high pressure Teflon bomb digestion. Approximately 300 mg of powdered certified reference material or real sample was placed in the TFM-PTFE vessel of the microwave system and moistened by 1 mL of 30% H2O2. Then, 4 mL of 65% HNO3 and 2 mL of 40% HF were added. The samples were heated for 20 min at 300 W. After microwave-assisted digestion, the clear solutions were transferred into 20-mL calibrated flasks and diluted to a volume with high-purity water. Prior to further analysis, these samples were appropriately diluted depending on the concentration level of Ag. A corresponding blank was also prepared according to the microwave-assisted digestion procedure detailed above.
Preconcentration and atomic absorption spectroscopy determination procedures. A total of 1000 mg of bentonite was placed into a 10-mL flask and filled with deionized water up to the mark to obtain a 10% (w/v) suspension. The bentonite was weighed using a microanalytical balance. The suspension of bentonite was dispersed for 1 min using a Sonopuls HD 70 ultrasonic cell disruptor/homogenizer equipped with a 2-mm titanium microtip. The sample solution (10 mL) was mixed with 5 mg of bentonite (as 50 μL of a 10% (w/v) suspension). The sample pH was adjusted to 8 using a pH-meter supplied with a combined glass electrode. Then, the sample was sonicated for 5 s using an ultrasonic homogenizer. Homogenization was achieved promoting a reaction between metal ions and bentonite. Subsequently, the mixture was centrifuged for 2 min at 4500 rpm. A centrifuge was employed for phase separation after the extraction procedure. Ag ions adsorbed on the bentonite settled at the bottom of the tube. Then, the aqueous phase was removed, and the metal-loaded bentonite was suspended using 0.2 mL of deionized water. To determine the analyte, 20 μL of the slurry was injected into the graphite tube for HR-CS GFAAS detection under optimized conditions. Before transfer to the graphite furnace, the samples were homogenized using the ultrasonic homogenizer for 5 s. Following DMSPE extraction, calibration was performed using the standard addition method. The extraction was carried out in three replicates for each sample (10 mL). After extraction, the liquid phase was removed, and the appropriate volumes of the standard solution were added to the metal-loaded bentonite. Next, distilled water was added to each sample to the volume of 0.2 mL to form a stable suspension before analysis.
The slurry should be stabilized using highly viscous liquid media to prevent rapid sedimentation [24]. In this study, the repeatability of the results obtained using ultrasonic homogenization of the slurry was satisfactory (relative standard deviation (RSD) ≤5%). Thereby, no slurry-stabilizing agent was added.
RESULTS AND DISCUSSION
Analytical blank. Slurry sample preparation minimizes the chance of sample contamination compared to conventional sample preparation procedures. However, there are still several possible sources of contamination, e.g., bentonite used as an adsorbent and the reagents used to optimize pH of the sample solution. Blank was determined using the same treatment procedure as for samples, i.e., DMSPE procedure. The absolute blank achieved for Ag was 0.004 ng.
Selection of instrumental conditions. The operating parameters of the HR-CS GFAAS instrument are given in Table 1. Silver was determined in its most sensitive wavelength (328.068 nm) as the primary resonance line. The peak areas of the absorbance signals were used for calculations. An analytical blank was also used throughout the procedure. The mean blank value was subtracted from the sample value after all calculations. During the study, the standard addition method was used for Ag determination.
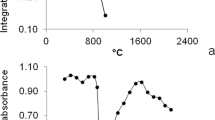
The temperature program was optimized for a standard solution containing 0.5 µg/L of Ag after preconcentration on bentonite. The influence of pyrolysis temperature on integrated absorbance was studied within the range of 450–850°C (Fig. 1) in the presence of a mixture of Pd(NO3)2 and Mg(NO3)2. Maximum analytical signal was achieved at a pyrolysis temperature of 650°C. After optimization of pyrolysis conditions, the effect of atomization temperature on Ag analytical signals was studied within the range of 1450–1950°C (Fig. 1). In this research, an optimum pyrolysis temperature of 650°C was used. Maximum analytical signal was achieved at an atomization temperature of 1750°C. The temperature program used for Ag determination in certified reference materials and environmental samples is shown in Table 2.

Optimization of pyrolysis and atomization temperatures performed for sample solution containing 0.5 µg/L of Ag after preconcentration on bentonite. Conditions: sample volume—10 mL, sample pH 8, centrifugation time—2 min, ultrasonication time—5 s, adsorbent amount—5 mg. The error bar is the standard deviation (n = 5).
Effect of sample pH. Sample pH plays an important role in the adsorption of Ag on bentonite. The oxides of aluminum, calcium, magnesium, iron, and silicon occur in bentonite. The hydroxylated surfaces of oxides develop a charge on the surface in an aqueous solution through amphoteric dissociation. The amphoteric oxides favor the adsorption of anions at lower pH, while adsorption of cations increases with increasing pH. The low adsorption of Ag+ on bentonite at lower pH is due to strong H+ ion competition for the available exchange sites [25].
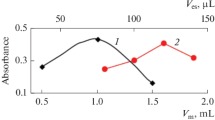
The influence of pH was investigated in the range of 3–10 (Fig. 2). Modification of pH was accomplished by adding appropriate amounts of NaOH or HNO3. The analytical signals remained low up to pH 4. It was observed that when pH was relatively low (4–7), integrated absorbance increased with pH. When the pH value was 8, the integrated absorbance of the analyte reached a maximum. However, when pH exceeded 8, the analytical signal decreased slightly for Ag. Therefore, in this study, the pH value of 8 was chosen for further experiments.

Influence of pH on Ag adsorption (0.5 µg/L) on bentonite. Conditions: sample volume—10 mL, centrifugation time—2 min, ultrasonication time—5 s, adsorbent amount—5 mg. The error bar is the standard deviation (n = 5).
Effect of bentonite amount. To obtain the highest analytical signals, an appropriate amount of bentonite as an adsorbent should be used in the DMSPE procedure. To achieve the optimum amount of bentonite necessary for quantitative enrichment of Ag, the introduced amount of adsorbent ranged from 1 to 15 mg per sample (10 mL). It was observed that the integrated absorbance increased with bentonite amount in the range of 1–5 mg (Fig. 3). This can be explained by the increase in the number of active sites on the bentonite surface available for binding metal ions. The analytical signal reached maximum at bentonite amount equal to 5 mg. The amount of bentonite was found to have a plateau of integrated absorbance in the range of 5–15 mg because the number of active sites was sufficient to bind all metal ions present in the samples. Therefore, in this study, 5 mg of bentonite was chosen for further experiments. The influence of the ultrasonication time on integrated absorbance of the analyte was also investigated within a range of 1–10 s, and the ultrasonication time of 5 s was chosen for further analysis.

Influence of bentonite amount on Ag adsorption (0.5 µg/L). Conditions: sample volume—10 mL, centrifugation time—2 min, ultrasonication time—5 s, sample pH 8. The error bar is the standard deviation (n = 5).
Adsorption capacity. To determine the adsorption capacity of bentonite, 20 mg of the adsorbent was added to 10 mL of the sample solution containing 25 mg/L of Ag. The experiment was performed at a pH value of 8. Following sonication for 10 s, the mixture was centrifuged for 3 min at 4500 rpm, and the water phase was collected. The concentration of remaining Ag ions in the water phase was determined using HR-CS GFAAS. The adsorption capacity (qt) was calculated from the Eq. (1):
where c0 and ct are the element concentrations before and after adsorption (mg/L), V is the sample volume (L), and W is the adsorbent mass (g) [10]. The adsorption capacity was found to be 8.5 mg/g for Ag.
Analytical figures of merit. In this work, the limit of detection (LOD) was calculated according to the International Union of Pure and Applied Chemistry (IUPAC) definition (Eq. (2)):
where SD is the standard deviation of 10 consecutive measurements of blank solution, and m is the slope of the addition graph [26]. The limit of detection was 0.01 µg/L for Ag.
Five replicate measurements of the total procedure blank solution were carried out, and the RSD of the background value for the raw data was calculated. The RSD was 5% for Ag. This reflects the precision of the total procedure. The absolute blank achieved for Ag was 0.004 ng. The linear range of the calibration curve was 0.05–5 µg/L.
The limit of detection achieved for Ag (0.01 µg/L) is seventy times better than LOD obtained using immobilized DDTC microcolumn [23], about eight times better than LOD obtained using silica-gel modified with 3-aminopropyltriethoxysilane [19], and two times better than LOD achieved using SPHERON® Thiol 1000 as a solid sorbent [22] prior to flame atomic absorption spectrometry. The limit of detection obtained using magnetic nanocomposite modified with thiourea [20] is superior by a factor of four to the limit of detection obtained in this work. The limit of detection reported by Avila et al. [21] is about thirty times better than that achieved for Ag using bentonite as a solid sorbent. The limit of detection obtained for Ag using alumina modified with polyethylenimine [18] is nine times better than LOD reported in this work.
While direct comparison of detection limits is often misleading owing to the use of different systems, operating conditions, and modes, it is clear that the detection limit that can be achieved using HR-CS GFAAS with slurry sampling after bentonite adsorption is better than this obtained for conventional GFAAS. The preconcentration factor achieved for Ag was 34. It was calculated using the ratio of the analyte concentration in the solid phase (c1) to the initial concentration of the analyte (c0) in the sample solution (PF = c1/c0) [27]. The liquid sample concentration used for assessing the detection limit and precision was 0.1 μg/L. A sample throughput is relatively high. Typically, at least 8 samples can be prepared within 30 min.
Accuracy verification. To ensure the accuracy and precision of the method, three certified reference materials were analyzed. These certified reference materials were chosen because they were the closest available to soil and water samples and are certified for the assessment of the analyte of interest. Results obtained for certified reference materials are summarized in Table 3. Short-term precision is expressed as the RSD of five replicate measurements of each sample. The results are in agreement with certified values for certified reference materials according to the t-test at a 95% confidence level. The obtained results show that the proposed method can be applied to the preconcentration and determination of Ag in environmental samples.
The certified reference materials used during the research have complicated matrices containing high concentrations (mg/kg) of elements such as Ba, Cu, Mn, P, S, and Ti, even at the % level (Al, Ca, Fe, K, Mg, Na, Si). The results of analysis obtained in the presence of these elements proved that the interferences from foreign ions can be ignored. For this reason, further studies of the matrix effect were not carried out.
Silver determination in environmental samples. To evaluate the usefulness of the proposed method for Ag determination, it was quantified in three water samples (seawater, lake water, and river water) and three soils collected in the vicinity of the Legnica Copper Smelter (South-West Poland, Silesia) using the experimental conditions optimized previously. The results for the samples analyzed using the proposed method are given in Table 4. Quantification of Ag was based on the standard addition method. In all cases, the precision (RSD) of replicate determination is approximately 5%.
REFERENCES
The Silver Institute. http://www.silverinstitute.org. Accessed October 11, 2021.
Abou El-Nour, K.M.M., Eftaiha, A., Al-Warthan, A., and Ammar, R.A.A., Arab. J. Chem., 2010, vol. 3, p. 135.
Kabata-Pendias, A. and Mukherjee, A.B., Trace Elements from Soil to Human, Heidelberg: Springer, 2007.
Nordberg, G.F., Fowler, B.A., Nordberg, M., and Friberg, L., Handbook of the Toxicology of Metals, San Diego: Elsevier, 2007, 3rd ed.
Vandecasteele, C. and Block, C.B., Modern Methods for Trace Element Determination, Chichester: Wiley, 1993.
Jiang, X., Huang, K., Deng, D., Xia, H., Hou, X., and Zheng, Ch., TrAC, Trends Anal. Chem., 2012, vol. 39, p. 38.
Atkovska, K., Bliznakovska, B., and Ruseska, G., J. Chem. Technol. Metall., 2016, vol. 51, p. 215.
Mohellebi, F. and Lakel, F., Desalin. Water Treat., 2016, vol. 57, p. 6051.
Melichova, Z. and Hromada, L., Pol. J. Environ. Stud., 2013, vol. 22, p. 457.
Hong, S., Wen, Ch., He, J., Gan, F., and Ho, Y.S., J. Hazard. Mater., 2009, vol. 167, p. 630.
Wang, S., Dong, Y., He, M., Chen, L., and Yu, X., Appl. Clay Sci., 2009, vol. 43, p. 164.
Krawczyk, M., Akbari, S., Jeszka-Skowron, M., Pajootan, E., and Fard, F.S., J. Anal. At. Spectrom., 2016, vol. 31, p. 1505.
Krawczyk-Coda, M., Spectrochim. Acta, Part B, 2017, vol. 129, p. 21.
Ghiasvand, A.R., Moradi, F., Sharghi, H., and Hasaninejad, A.R., Anal. Sci., 2005, vol. 21, p. 387.
Hartmann, G., Hutterera, C., and Schuster, M., J. Anal. At. Spectrom., 2013, vol. 28, p. 567.
Fathabad, S.D. and Hasanvandi, S., Orient. J. Chem., 2012, vol. 28, p. 1311.
Ghiasvand, A., Shadabi, S., Hajipour, S., and Nasirian, A., Anal. Bioanal. Chem. Res., 2015, vol. 2, p. 60.
Taher, M.A., Daliri, Z., and Fazelirad, H., Chin. Chem. Lett., 2014, vol. 25, p. 649.
Ekinci, C. and Köklü, Ü., Spectrochim. Acta, Part B, 2000, vol. 55, p. 1491.
Ghanei-Motlagh, M., Fayazi, M., Taher, M.A., and Jalalinejad, A., Chem. Eng. J., 2016, vol. 290, p. 53.
Avila, A.K. and Curtius, A.J., J. Anal. At. Spectrom., 1994, vol. 9, p. 543.
Medved, J., Matúš, P., Bujdoš, M., and Kubová, J., Chem. Pap., 2006, vol. 60, p. 27.
Dadfarnia, S., Haji Shabani, A.M., and Gohari, M., Talanta, 2004, vol. 64, p. 682.
Kurfürst, U., Solid Sample Analysis: Direct and Slurry Sampling Using GF-AAS and ETV-ICP, Heidelberg: Springer, 1998.
Khan, S.A., Rehman, R., and Khan, M.A., Waste Manage., 1995, vol. 15, p. 271.
Currie, L.A., Pure Appl. Chem., 1995, vol. 67, p. 1699.
Rydberg, J., Solvent Extraction Principles and Practice, Revised and Expanded, Boca Raton: CRC Press, 2004, 2nd ed.
Funding
This work was supported the Ministry of Education and Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Krawczyk-Coda, M. Determination of Silver in Environmental Samples by High-resolution Continuum Source Graphite Furnace Atomic Absorption Spectrometry after Preconcentration on Bentonite. J Anal Chem 77, 1155–1161 (2022). https://doi.org/10.1134/S1061934822090076
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934822090076