
Abstract
We developed a method for determining ethyl methylphosphonic, isopropyl methylphosphonic, isobutyl methylphosphonic, and pinacolyl methylphosphonic acids in human urine using gas chromatography with high-resolution mass spectrometry and preliminary derivatization with p-methoxyphenacyl bromide. All of the acids are specific hydrolysis products and biomarkers of nerve agents. Conditions of the derivatization reaction were optimized. Features and general patterns of the MS1 and MS2 spectra of derivatives acquired in electron and chemical ionization modes are revealed. The developed method achieved the limits of detection in the range from 20 to 70 ng/mL.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Chemical weapons (CW), a damaging element of “mass destruction weapons,” were widely used in hostilities during the First World War, and by this day, some of the CW remain in storage even after mass destruction following the Chemical Weapons Convention [1]. The most toxic nerve agents, such as sarin, soman, VX, and VR, are well known and continue to be exploited by terrorists.
After exposure, nerve agents are rapidly distributed and metabolized in a human body, forming free metabolites and covalent adducts with macromolecules, such as enzymes and other proteins [2]. Among these compounds, there are specific biomarkers of exposure to CW, because there are no known alternative routes for their entry into a living organism [3]. The level of such metabolites and adducts in biological fluids may reflect the degree of exposure to CW. The determination of metabolites excreted with urine is one of the most convenient ways to confirm the effect of CW on a body [3]. The main metabolites of nerve agents in urine are hydrolysis products, namely, alkyl methylphosphonic acids (AMPAs) (Fig. 1). For example, sarin (GB), soman (GD), VX, and VR are hydrolyzed to isopropyl methylphosphonic acid (iPrMPA), pinacolyl methylphosphonic acid (PinMPA), ethyl methylphosphonic acid (EMPA), and isobutyl methylphosphonic acid (iBuMPA), respectively. These AMPAs are ultimately hydrolyzed to methylphosphonic acid (MPA). Several analytical approaches have been developed for the identification of AMPAs as biomarkers of exposure to nerve agents. Gas chromatography–mass spectrometry (GC–MS) [4–6], gas chromatography–tandem mass spectrometry (GC–MS/MS) [7–11], and liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) [12–15] are the most frequently used methods because of their high sensitivity and selectivity. According to the analysis quality control system adopted by the Organization for the Prohibition of Chemical Weapons (OPCW), CW markers must be reliably identified, which can be achieved through the use of gas chromatography with high-resolution mass spectrometry (GC–HRMS), the accuracy of mass measurement of which is no more than 5 ppm.

Hydrolysis of nerve agents.
Alkyl methylphosphonic acids are polar and low-volatile compounds; therefore, samples preparation of biological samples for GC-based analysis is usually based on the preliminary recovery of the analytes by solid-phase extraction (SPE) and the subsequent derivatization. Several SPE procedures have been used to determine AMPAs in biomedical matrices. Some included anion-exchange SPE [4, 6, 10] or polymer-reversed-phase SPE [5], but few are reasonably reproducible, probably because of changes in the SPE cartridge composition combined with the variability inherent to urine [9]. Derivatization of AMPAs in biological samples is usually achieved by methylation [7, 8], trimethylsilylation [4], tert-butyldimethylsilylation [6], or pentafluorobenzylation [9, 10]. Methylated and silylated derivatives can be analyzed by GC–MS and GC–MS/MS with positive ion detection. However, derivatization with p-methoxyphenacyl bromide (PMPB) is better suited for the highly sensitive determination of AMPAs [16], because ester derivatives have excellent gas chromatographic properties and good ionizability, which makes them easily detectable at low concentrations using mass spectrometry in the positive ion detection mode. The derivatization reactions leading to the blocking of the hydroxyl groups of AMPAs increase the molecular weight and decrease the polarity of the compounds, resulting in significantly lower limits of detection and improved performance of determination by both GC–MS and HPLC–MS.
This work aimed to study possibilities of using p‑methoxyphenacyl bromide as a reagent for the preparation of derivatives of the highest priority alkyl methylphosphonic acids in the sample preparation of biomedical samples for GC–high-resolution MS analysis.
EXPERIMENTAL
Solutions and reagents. We used the following reagents: ethyl methylphosphonic acid (EMPA), isopropyl methylphosphonic acid (iPrMPA), isobutyl methylphosphonic acid (iBuMPA), and pinacolyl methylphosphonic acid (PinMPA), all of a purity of >98% (Sigma, Aldrich, United States), p-methoxyphenacyl bromide (PMPB) (>98%, Sigma Aldrich, United States), potassium carbonate (>99%, Merck, Germany), triethylamine (>99%, Acros Organics, United States), acetonitrile (HPLC grade, Panreac, Spain), and deionized water purified using a Milli-Q system (Millipore, United States).
Preparation of stock solutions. Standard samples of EMPA, iPrMPA, iBuMPA, and PinMPA were weighed and dissolved in acetonitrile to prepare concentrated stock solutions (1 mg/mL) of each compound; the solutions were stored at 4°C. A working solution containing each analyte in a concentration of 200 μg/mL was prepared by mixing equal volumes of individual stock solutions in acetonitrile. Seven standard calibration solutions (50, 100, 200, 400, 500, and 1000 ng/mL) were prepared by adding a diluted working solution to human urine, which initially contained no test substances.
Derivatization of AMPAs with p-methoxyphenacyl bromide. Additives of AMPA solutions were added to urine samples (1 mL) and diluted with 9 mL of acetonitrile. The resulting solution was centrifuged for 3 min at 14 000 rpm and loaded onto a SampliQ SILICA cartridge (Agilent Technologies, 100 mg, 1 mL), previously activated with 1 mL of an acetonitrile–water mixture (75 : 25, vol) and 2 mL of acetonitrile. After passing the analyte solution, the cartridge was washed with acetonitrile (1 mL). The target analytes were eluted with an acetonitrile–water mixture (75 : 25, vol) (2 × 1 mL). The eluate was preconcentrated at 80°C under nitrogen. The dry residue was dissolved in 100 μL of acetonitrile containing 10 mg/mL of PMPB. Triethylamine (50 mg) was added to the resulting solution, and the mixture was kept at 80°C for 40 min.
Conditions for the GC–MS determination of the AMPA derivatization products. We used a 7890A gas chromatograph (Agilent Technologies, United States) in combination with a Q-TOF 7200 high-resolution mass spectrometer (HRMS) equipped with an electron and a chemical ionization source (Agilent Technologies, United States). Chromatographic separation was performed in a quartz capillary column with a DB-5MS stationary liquid phase with the length 30 m, internal diameter 0.25 mm, and the phase thickness 0.25 μm. Separation was carried out in the temperature programming mode: 40°C for 1 min, heating from 40 to 280°C at a rate of 30oC/min, 280°C for 25 min (electron ionization), 40°C for 1 min, heating from 40 to 280°C at a rate of 10oC/min, 280°С for 10 min (chemical ionization). The injector temperature was 250°C; the detector interface temperature was 290°C. Helium was used as a carrier gas at a flow rate of 1 mL/min. The injected sample volume was 1 μL.
Mass spectrometric detection was carried out in the electron ionization (EI) and chemical ionization (CI) modes with the following parameters. EI mode (70 eV): ion source temperature 230°C, mass filter temperature 150°C, scanned mass range from 40 to 500 Da, scanning frequency 10 000 scan s–1; CI mode with detection of positively charged ions (240 eV): ion source temperature 150°C, mass filter temperature 100°C, scanned mass range from 60 to 500 Da, scan rate 10 000 scan s–1, reagent gas methane with a feed rate of 20% for the detection of positively charged molecules. The width of the isolation window of the precursor ions is 0.1 Da; the gas in the collision cell is nitrogen with a feed rate of 1.5 mL/min. To detect the products of AMPA derivatization with PMPB under EI conditions, the following selective transitions with their optimal collision energies were used: EMPA-PMPB m/z 272 → m/z 135.0446 (5 eV), iPrMPA-PMPB m/z 286 → m/z 135.0446 (5 eV), iBuMPA-PMPB m/z 300 → m/z 135.0446 (10 eV), PinMPA-PMPB m/z 328 → m/z 135.0446 (10 eV); under CI conditions: EMPA-PMPB m/z 245 → m/z 149.0590 (1 eV), iPrMPA-PMPB m/z 245 → m/z 149.0590 (1 eV), iBuMPA-PMPB m/z 245 → m/z 149.0590 (1 eV), PinMPA-PMPB m/z 245 → m/z 149.0590 (1 eV) (Table 1).
RESULTS AND DISCUSSION
Derivatization. The interaction of AMPAs with PMPB may proceed by the reactions shown in Scheme 1. The derivatization reaction is a SN2 process, proceeding by a nucleophilic attack of the methylene group of PMPB on the AMPA anion. The reaction requires an alkaline medium, so that AMPAs dissociate to form an active anion. To create an alkaline medium, we used organic and inorganic bases: triethylamine and potassium carbonate. The α-position of the carbonyl group is preferred for SN2 processes; therefore, the substitution of bromine proceeds efficiently, and the reaction yield is relatively high.

Scheme 1 .
We studied the effect of various types of bases, temperature and time of the reaction, and the extractant on the derivatization yield. The best yields and low noise were obtained using trimethylamine, compared to the other base tested (potassium carbonate). Acetonitrile was used as a solvent for derivatization, according to [16]. The study of the effect of temperature showed that when exceeding 80°C, a significant increase in yields was not observed. This is because an increase in the reaction temperature also accelerated the side (interfering) processes; the optimal reaction temperature was set at 80°C. The reaction time was varied from 0.5 to 12 h; the time of 40 min was selected the optimal.
Extraction of AMPAs from urine using SPE. Several SPE cartridges were tested to remove interfering components from the sample and to improve the yield of AMPA-PMPB, including SampliQ SILICA (Agilent Technologies, 100 mg, 1 mL), OAZIS HLB (Waters, 30 mg, 1 mL), and SiOH (Macherey-Nagel, 100 mg, 1 mL). To calculate the recovery of analytes, two additions of the AMPA working solution (1000 ng/mL) were made: one to the blank urine samples before passing through the SPE cartridges and another to the eluates obtained after passing the blank urine containing no target analytes. After that, evaporation and derivatization were carried out, followed by the gas chromatographic separation of the resulting mixture of analytes (Fig. 2). The recovery was calculated using the following equation: R = (S1/S2) × 100, where S1 and S2 are the areas of AMPA-PMPB peaks in samples with the addition of analytes before and after SPE, respectively. The recovery of AMPAs are presented in Table 2.

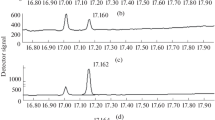
Selected ion chromatograms of the urine samples with an AMPA concentration of 1000 ng/mL (a) in the EI (m/z 135.0446) and (b) CI (m/z 245.0570) modes. PinMPA-PMPB ester generates two peaks due to the chromatographic separation of two pairs of diastereoisomers. The range of scanned m/z is from 40 to 500.
Table 2 shows that the highest analyte recovery were obtained using SILICA cartridges, which were used for further experiments.
Features of the mass spectra of AMPA derivatives under chemical and electron ionization conditions. Molecular ions were detected in the mass spectra of AMPA derivatives (Fig. 3a) under EI conditions. In the CI mode with the detection of positively charged ions for all AMPA derivatives, we detected protonated molecules (Fig. 3b), which, however, also underwent collision-induced dissociation (CID) with the formation of a specific fragment ion with m/z 245.0570, also observed in the mass spectrum of the MPA derivative.

(a) EI and (b) CI mass spectra of EMPA-PMPB, iPrMPA-PMPB, iBuMPA-PMPB, and PinMPA-PMPB.
Next, we obtained MS2 spectra of product ions for all AMPA derivatives using CID. The signals with m/z 149 and 121 were the most intense in the mass spectra of almost all the test compounds (Scheme 2); they correspond to positively charged product ions included in the reagent structure. In the EI mode, the ion peak with m/z 135 was the most intense in the mass spectra of product ions.

Scheme 2 .

Chromatograms and MS2 spectra of the urine samples with an iPrMPA-PMPB concentration of 100 ng/mL (a, b) in the EI mode m/z 286 → m/z 135.0446 (5 eV) and (c, d) in the CI mode m/z 245 → m/z 149.0590 (1 eV).
Validation of the developed method. The limit of detection (LOD) was determined as the concentration at which the ratio of the signal of the test sample to the noise of the blank sample is 3 : 1. According to the concentration LOD values obtained by the two methods (Table 3), we can conclude that the developed method is sufficiently sensitive and reliable, since tandem HRMS transitions in the electron and chemical ionization modes are used.
The accuracy of the developed procedure was confirmed by the standard addition method (Table 4). For this, AMPAs were added to urine samples at three concentration levels. The added and found values practically coincided, which indicates the high reliability of the developed method.
CONCLUSIONS
We developed a simple, sensitive, and reliable method for determining the metabolites of nerve agents in human urine. High sensitivity and selectivity were achieved using gas chromatography–high-resolution mass spectrometry in chemical and electron ionization modes. The MS1 and MS2 spectra of the derivatives of the test compounds, obtained using CI and EI, were recorded, and the features and patterns of their formation were shown. We selected characteristic ions that enable reliable identification and sensitive determination using selective ion transitions based on the interpretation of mass spectra. The conditions for the chromatographic separation of analytes were found, which yield good resolution in an adequate analysis time. The limits of detection by the developed procedure were in the range from 20 to 70 ng/mL for all four AMPAs. The proposed procedure can help to diagnose the effect of nerve-paralytic organophosphates.
REFERENCES
Organisation for the Prohibition of Chemical Weapons. http://www.opcw.org. Accessed June 5, 2020.
Black, R.M. and Noort, D., in Chemical Weapons Convention Chemicals Analysis: Sample Collection, Preparation and Analytical Methods, Mesilaakso, M., Ed., Chichester: Wiley, 2005, p. 403.
Black, R.M., J. Anal. Toxicol., 2008, vol. 32, no. 1, p. 2.
Wang, Q.Q., Xie, J.W., Gu, M.S., et al., Chromatographia, 2005, vol. 62, no. 3, p. 167.
Shih, M.L., Smith, J.R., McMonagle, J.D., et al., Biol. Mass Spectrom., 1991, vol. 20, no. 11, p. 717.
Kataoka, M. and Seto, Y., J. Chromatogr. B: Biomed. Sci. Appl., 2003, vol. 795, no. 1, p. 123.
Driskell, W.J., Shih, M., Needham, L.L., et al., J. Anal. Toxicol., 2002, vol. 26, no. 1, p. 6.
Barr, J.R., Driskell, W.J., Aston, L.S., et al., J. Anal. Toxicol., 2004, vol. 28, no. 5, p. 372.
Riches, J., Morton, I., Read, R.W., et al., J. Chromatogr. B: Biomed. Sci. Appl., 2005, vol. 816, nos. 1–2, p. 251.
Fredriksson, S.A., Hammarstrom, L.G., Henriksson, L., et al., J. Mass Spectrom., 1995, vol. 30, no. 8, p. 1133.
Subramaniam, R., Ostin, A., Nilsson, C., et al., J. Chromatogr. B: Biomed. Sci. Appl., 2013, vol. 928, p. 98.
Ciner, F.L., McCord, C.E., and Plunkett, R.W., Jr., et al., J. Chromatogr. B: Biomed. Sci. Appl., 2007, vol. 846, nos. 1–2, p. 42.
Evans, R.A., Jakubowski, E.M., Muse, W.T., et al., J. Anal. Toxicol., 2008, vol. 32, no. 1, p. 78.
Mawhinney, D.B., Hamelin, E.I., Fraser, R., et al., J. Chromatogr. B: Biomed. Sci. Appl., 2007, vol. 852, nos. 1–2, p. 235.
Baygildiev, T., Zatirakha, A., Rodin, I., et al., J. Chromatogr. B: Biomed. Sci. Appl., 2017, vol. 1058, p. 32.
Baygildiev, T.M., Vokuev, M.F., Oreshkin, D.V., et al., J. Anal. Chem., 2020, vol. 75, p. 1708.
Funding
The study was supported by the Russian Foundation for Basic Research, project no. 18-33-20068 mol_a_ved.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by O. Zhukova
Rights and permissions
About this article

Cite this article
Oreshkin, D.V., Baygildiev, T.M., Vokuev, M.F. et al. Determination of p-Methoxyphenacyl Bromide Derivatives of Alkylmethylphosphonic Acids in Urine Using Gas Chromatography with High-Resolution Mass Spectrometric Detection. J Anal Chem 76, 1530–1537 (2021). https://doi.org/10.1134/S1061934821130098
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934821130098