
Abstract
Conditions of the synthesis of nanoparticles of ractopamine molecularly imprinted polymers (MIP) by the precipitation method are studied. It is shown that the size and dispersity of MIP particles are largely determined by the nature of the functional and cross-monomers and by the synthesis conditions: temperature, mixing rate, duration of synthesis, and ultrasonic treatment of the polymerization mixture before and after the synthesis. It is found that ractopamine MIP particles based on methacrylic acid and ethylene glycol dimethacrylate, sonicated for 30 min, have a minimum size and degree of dispersion. Using the piezoelectric quartz micro-weighing method, characteristics of the recognition layer based on MIP nanoparticles are found, and the analytical characteristics of a sensor for the determination of ractopamine in an aqueous solution are calculated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Molecularly imprinted polymers (MIP) have already have a good history as highly selective sorbents capable of replacing natural antibodies in many cases of the development of diagnostic tools for the sensitive recognition and determination of various chemical compounds. Unlike biomolecules, MIP are stable in the range of low and high pH values, on changes in pressure and temperature, have lower cost, the reaction of their formation proceeds more easily, and they can be synthesized for a wide range of substances and used both in organic and in water media. It was noted that imprinted polymers of regular shape have the best properties, especially in the nanoscale region [1], which is explained by a higher value of the ratio of their surface area to the volume. This improves the availability of surface imprints for analyte molecules and contributes to an increase in the rate of re-binding [2]. In addition, MIP nanoparticles easily remain in solution and are easier to dose [3], which creates prerequisites for their widespread use in sensors of various nature as substitutes for antibodies in pseudoimmunoassay [4–7].
The most popular methods for the synthesis of MIP nanoparticles are micro- and miniemulsion polymerization [8, 9], precipitation [10–14], and “core–shell” synthesis (core–shell, core–shell by grafting) [15–18] (analytical applications of MIP nanoparticles obtained by such methods were described in [19, 20]).
A convenient and simple precipitation method results in the production of spherical MIP particles with a high degree of homogeneity. In this method, polymerization takes place in the presence of a larger amount of a porogenic solvent. Individually growing polymer chains capture oligomers, monomers, and template molecules from a solution without coagulating or sticking together, and deposition takes place only when a critical size is achieved under the influence of gravitational forces. By controlling the polymer solubility parameters and solvent polarity, tone can obtain particles with constant structural pores and constant average sizes in the range from 0.01 to 1 μm, as well as with high yields (>85%) [5, 10]. The polymer spheres obtained by the precipitation method because of the cross-linking of linear chains with a cross-monomer are protected from aggregation due to surface tension forces. The diameter of MIP nanospheres obtained by the precipitation method can be reduced by the ultrasonic treatment of the polymerization mixture.
Despite the obvious advantages of the MIP nanospheres as substitutes for antibodies, the development of sensors based on them has began relatively recently; the most actively developing are piezoelectric sensors [21]. In particular, MIP nanoparticles have already proven themselves positively as recognition elements in piezoelectric sensors [22–24]. However, piezoelectric sensors based on MIP nanospheres were not previously used to determine ractopamine.
Ractopamine (Ract) is a non-specific beta-adrenergic receptor agonist and is used in animal husbandry in some countries as a stimulator of muscle growth, which results in its accumulation in tissues and fatty layers and causes toxic reactions, tachycardia, and muscle pain in people consumed such products; negatively affects the sexual development of children and the endocrine balance in a body. In accordance with the decision of the Codex Alimentarius Commission, the maximum permissible concentration (MPC) of ractopamine in meat (pork and beef) was set at 0.01 mg/kg, in the liver—0.04 mg/kg, and in the kidneys—0.09 mg/kg [25, 26]. Despite the proposal by the FAO/WHO Joint Expert Committee on Food Additives (JECFA) to establish the maximum residual ractopamine levels for pork and cattle, ractopamine is not approved for use in livestock and poultry farming in Russia, the EU, and China, and the import of products containing ractopamine is completely prohibited. However, ractopamine can still often be found in pork and feed.
The known instrumental methods of analysis do not provide sufficient sensitivity (photometry), are prolong, and require the use of expensive equipment (gas and liquid chromatography, capillary electrophoresis). Such methods are of little use for routine analyzes of a large number of food samples. It is necessary to develop simple and highly sensitive methods for the rapid determination of residual amounts of ractopamine in food products.
In recent years, in world practice, rapid enzyme immunoassay methods, immunosensors (mainly electrochemical and based on surface plasmon resonance), and MIP-based sensors have actively been used to control ractopamine in food products [27–29]. However, a high degree of crosslinking of molecularly imprinted systems impedes the transfer of electrons and photons and results in a decrease in the sensitivity and accuracy of the detection of analytes by electrochemical and optical sensors. MIP-based piezoelectric gravimetric sensors combine the advantages of molecular imprinting and piezoelectric detection methods and are, therefore, developed more actively. A piezoelectric sensor based on silanes molecularly imprinted by ractopamine was developed [30], but nanoparticles of ractopamine MIP obtained by the precipitation method were not previously used in piezoelectric sensors.
The aim of this work was to study the synthesis conditions of ractopamine MIP nanoparticles by the precipitation method and the formation of a recognizing coating of a piezoelectric sensor for the determination of ractopamine in liquid media.
EXPERIMENTAL
Materials and equipment. The following reagents were used to synthesize MIP nanoparticles: methacrylic acid (MA), methyl methacrylate (MMA), 4-vinylpyridine (4-VP) (Sigma-Aldrich, Great Britain) as a functional monomer; cross-monomer ‒ ethylene glycol dimethacrylate (EGDMA) (Vekton, Russia), divinylbenzene (DVB), trimethylpropane trimethacrylate (TPTA) (Sigma-Aldrich, Great Britain); polymerization initiator ‒ azobisisobutyronitrile (Labtekh, Russia); and template ‒ ractopamine hydrochloride (Sigma-Aldrich, Great Britain). Salbutamol (Sigma-Aldrich, Great Britain), clenbuterol hydrochloride, (Abcam, Great Britain), penicillin G, (Biochemist, Russia), and cefotaxime (Borisov Plant of Medicines, Russia) were used to study the sensor cross-reaction. Acetonitrile, toluene (Vekton, Russia), tetrahydrofuran (Sigma-Aldrich, Great Britain), ethyl alcohol of the reagent grade (Metafraks, Russia), and conc. CH3COOH (Base No. 1 of chemical reagents, Russia) were used as solvents. Cyanoacrylate was used for immobilization.
The synthesis of nanoparticles of ractopamine molecularly imprinted polymer by the precipitation method was carried out in a round-bottom flask equipped with a mechanical stirrer. Ractopamine chloride, a functional monomer, and a cross-monomer, taken in a ratio of 1 : 1 : 5, were dissolved in 25 mL of a porogenic solvent acetonitrile–toluene (4 : 1, vol). The prepolymerization mixture was sonicated for 2 min (PSB-2835-03 ultrasonic bath; PSB-Hals, Russia) at a frequency of 40 kHz, transferred to a thermostatic water bath, a polymerization initiator was added, and polymerization was carried out at constant stirring at a rate of 30 rpm. After the synthesis was completed, the colloidal solution of MIP particles was sonicated for 15 min.
The synthesis of non-imprinted polymer (NIP) nanoparticles was carried out by a similar procedure without ractopamine.
To study the laws of the polymerization process and the properties of the imprinted polymer, the data obtained by piezoelectric quartz weighing, infrared spectroscopy (Shimadzu Iraffinity-1 IR spectrometer, Japan), and optical microscopy (DigiMicro Skale microscope, DNT, Germany) were used. The size of the MIP and NIP nanospheres was determined using a Solver P47-PRO scanning probe microscope (Nanotechnology-MDT, Russia) in the tapping mode in air at a scanning rate of 1.34 Hz, using cantilevers NSGO 1/20 made of silicon with a nominal hardness of 5 N/m, with resonant scanning frequency in the range 120–180 kHz and radius of curvature 10 nm. Using the Nova software including the Grain Analysis function, the sizes of individual MIP particles localized in different parts of a graphite substrate were determined. Particle dispersity was estimated by the value of the confidence interval of the average diameter calculated for each batch of MIP nanoparticles (Fig. 1).

AFM images of particles of molecularly imprinted polymer methacrylic acid–ethylene glycol dimethacrylate.
Immobilization of molecularly imprinted polymer nanoparticles on the surface of a sensor electrode. Six milligrams of MIP particles (until the template was removed) were added to 5 mL of a cyanoacrylate solution (2 mg/mL) in tetrahydrofuran. The resulting mixture was homogenized in an ultrasonic bath for 3 min. Then, 2 mL of the obtained suspension was applied as a drop to the center of a piezoelectric crystal resonator rotating at a speed of 900 rpm, previously degreased with acetone and washed with acetonitrile, and MIP particles were immobilized by the spin-coating method [31] for 1–2 min by controlling the uniformity of the layer by optical microscopy. The suspension application was repeated until a uniform layer was obtained.
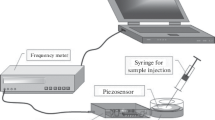
AT-cut piezoelectric quartz resonators with a natural oscillation frequency of 10 MHz, 8 mm in diameter with 5-mm gold electrodes on both sides (ETNA, Russia) were used as sensors. To measure the analytical signal, a setup included a B5-30 oscillation generator and a digital Diskop module (Bafika, Russia) connected to a personal computer was used. The analytical signal of the sensor was measured in a batch mode after drying the coating to a constant weight (dip & dry method).
To remove template molecules from the surface of MIP particles immobilized on the electrode, the sensor was immersed in 75 mL of a regenerating solution and kept at constant stirring for a certain time. Then it was dried under a stream of warm air to a constant weight twice-distilled water (solution 1), a 25% solution of acetic acid (solution 2), and a mixture of ethyl alcohol with acetic acid (25%) (9 : 1, vol) (solution 3) were used as a regenerating solution.
The recognition layer based on MIP nanoparticles was characterized by piezoelectric quartz microweighting similar to that described in [22, 31]. The analytical signal was the change in oscillation frequency after drying the sensor to a constant mass in air before and after the formation of a film based on MIP nanoparticles, after incorporation into molecular imprints and the removal of ractopamine molecules.
The mass of the polymer coating of the sensor m was calculated by the Sauerbrey equation: ΔF = ‒2.3 × \({\text{1}}{{0}^{{\text{6}}}}{{F_{0}^{{\text{2}}}m} \mathord{\left/ {\vphantom {{F_{0}^{{\text{2}}}m} S}} \right. \kern-0em} S},\) where S was the area of the electrode of the piezoelectric quartz sensor (0.10 cm2), F0 was resonator basic frequency, Hz. The analytical signal of the sensor was measured relative to a reference sensor based on the NIP: ∆Fan = ∆FMIP ‒ ∆FNIP, the mass of the extracted (ΔmMIP) and re-embedded ractopamine (ΔmREM(Ract) and Δ\(m_{{{\text{REM(Ract)}}}}^{'}\)) was determined by the difference between the mass of the MIP layer before and after template extraction; the concentration of surface ractopamine imprints on the surface of a layer of MIP nanoparticles (Rmax) and imprints after surface regeneration (R') was calculated as R = mEM/(SMRact), μmol/cm2 [31]. The selectivity of the recognition layer was evaluated by cross-reaction coefficients: CR, % = ΔFint/ΔFRact.
RESULTS AND DISCUSSION
Synthesis conditions for nanoparticles of ractopamine molecularly imprinted polymer. The size, dispersity, and the degree of imprinting of MIP particles are largely determined by the nature of functional and cross-monomers, porogenic solvent, and synthesis conditions: temperature, mixing rate, duration of synthesis, and ultrasonic treatment of the polymerization mixture before and after the synthesis. The effective non-covalent interaction between the template and the functional monomer, as well as the stability of the prepolymerization complex determines the affinity of the MIP and the selectivity of the subsequent recognition of template molecules. It was previously noted [23] that the best results in the synthesis of MIP nanospheres by the precipitation method were achieved when MA, MMA, and 4-VP were used as functional monomers, because of their ability to form hydrogen bonds with molecules of various templates, and EGDMA, TPTA, and DVB, which ensure the stability of the polymer matrix and imprinting sites, as well as affect the size of MIP nanoparticles and their number were used as cross-monomers. To initiate the process of free radical polymerization, azobisisobutyronitrile was selected as the initiator; it works efficiently in the range 60–75°C and is highly soluble in a porogenic solvent. In choosing a solvent, the solubility of all components in it was taken into account. Previous studies have shown that the use of an acetonitrile–toluene mixture (4 : 1, vol) resulted in particles of the minimum size.
In the course of the experiment, MIP ractopamine particles based on MA, 4-VP, and MMA were obtained using EGDMA, TPTA, and DVB as cross-reactants (Table 1).
The results of atomic force microscopy (AFM) studies showed that MIP particles based on MA–EGDMA had the minimum diameter (Fig. 1), in contrast to MIP particles based on 4-VP–EGDMA (Fig. 2a), which were located on the plane in scanning as chains and various clusters. In addition, such particles were unstable for the further use. They dissociated into polydisperse aggregates when they were dissolved in tetrahydrofuran being immobilized on the surface of the sensor electrode. MIP particles based on MMA–EGDMA (Fig. 2b) were large conglomerates of irregular shape with a high degree of dispersion, which complicated their further use in piezoelectric sensors as “artificial antibodies.”

AFM images of particles of molecularly imprinted polymer (a) 4-vinylpyridine–ethylene glycol dimethacrylate and (b) methacrylic acid–ethylene glycol dimethacrylate.
In the synthesis of MA-based MIP particles using EGDMA, DVB, and TPTA, monodisperse nanospheres formed only in the first case, whereas in using DVB and TPTA, the particles had gel-like structures (as droplets, bulges on the substrate surface). The presence of three alkyl groups in the TPTA molecule and two vinyl groups in the DVB molecule contributes to obtaining a looser MIP structure, which facilitates binding of template molecules; however, this simultaneously results in an excessive increase in the number of surface prints and particle size, which limits their use as “artificial antibodies”. At the same time, the use of EGDMA as a cross-reagent facilitates the synthesis of particles of a minimum size due to the formation of shorter linkers.
The size of the synthesized particles can be controlled by changing mixing rate, the duration of the synthesis, and the temperature of the prepolymerization mixture. The synthesis of MIP particles of ractopamine based on MA–EGDMA with stirring at a rate of 30, 100, and 250 rpm showed that an increase in the stirring rate resulted not only in an increase in the diameter of MIP particles by 45–50 times, but also to the formation of a gel or loose particles of irregular shape, compared with particles obtained at a rate of 30 rpm. Similarly, the diameter of the synthesized MIP particles is affected by an increase in the duration of synthesis from 3 to 6 h, which is explained by the formation of a larger number of oligomers and, therefore, the absorption of high concentrations of functional and cross-monomers. At that, the MA–EGDMA particle size significantly increased from 34 ± 5 to 650 ± 55 nm, as well as polydispersity.
By changing the temperature of the polymerization mixture, it was possible to influence the rates of individual stages of synthesis and the stability of the prepolymerization complex by increasing or decreasing the kinetic energy of the system [32]. In the experiment, MIP particles of ractopamine based on MA–EGDMA were obtained at 400, 600, and 800°C. Using AFM scanning, it was found that, at 400°C, stable monodisperse spherical MIP particles could not be obtained because of the low polymerization rate in the same time period (3 h), which undoubtedly affected the structure of MIP particles. At 800°C, MIP particles were obtained in a “dry” form, in the form of a “powder,” in contrast to all other polymer samples obtained in suspensions. The particles obtained at this temperature have regular, clear, and spherical shapes; however, their drawbacks were the relatively large diameter compared to particles obtained at 600°C and their relative polydispersity: the diameter of individual spheres, reached 2 μm.
It is known that the use of ultrasound after the completion of synthesis significantly reduces particle size and polydispersity, and the preliminary processing of the prepolymerization mixture with ultrasound helps to remove oxygen from it, which inhibits the polymerization of MIP particles. In the experiment, the duration of ultrasonic treatment of the prepolymerization mixture was increased from 2 to 15 min, and the effect of ultrasound on the size of the formed MA–EGDMA particles was investigated. It was shown that an increase in the duration of ultrasonic treatment of the prepolymerization mixture to 15 min resulted in the formation of larger MIP particles and a loose inhomogeneous structure compared to particles obtained in processing for 2 min. Ultrasonic treatment of MIP particles after synthesis, on the contrary, significantly reduced particle size (Table 2).
Study of the structure of nanoparticles of ractopamine molecularly imprinted polymer by IR spectroscopy. To reveal the nature of bonds formed in the synthesis of MIP, to establish functional groups involved in nonspecific interactions with the template molecules, IR spectra of the functional monomer (MA) and ractopamine were compared with the spectra of MIP and NIP. The broadening and shift of the peak of methacrylic acid carbonyl groups to the short-wavelength region in the MIP spectra from 1697 to 1728 cm–1, as well as the broadening of the peaks of hydroxyl groups (945–941 cm–1) indicate their participation in the formation of hydrogen bonds with the ractopamine molecule in the formation of a prepolymerization complex (Fig. 3). A similar shift and broadening of the peak of the –COOH group in the NIP spectrum can point to the participation of the carbonyl group not only in the interaction with ractopamine, but also in the polymerization.

IR spectra of methacrylic acid, ractopamine, molecularly imprinted polymer, and non-imprinted polymer.
In the spectra of MIP and ractopamine there was a band of the –C–OH alcohol group (1056–1053 cm–1), which was absent in the NIP spectrum, pointing to the presence of ractopamine in the polymer matrix of the imprinted particles. The broadening of the –C–OH band in the MIP spectrum confirms the participation of hydroxyl groups in the formation of hydrogen bonds with the functional monomer. Only in the IR spectrum of MIP, the bands of the –NH– group (1512 cm–1) and of the 1,4-substituted aromatic (phenolic) group –C6H4–OH (1220–1170 cm–1), which are characteristic of ractopamine, were observed. A shift of the vibrational frequency of the –NH– group to the short-wave region in the polymer compared to pure ractopamine indicates its participation in the formation of hydrogen bonds. In polymerization in the presence of a cross-monomer, complexes of monomers with the template are fixed in certain positions of a rigid polymer structure due to the formation of cross-links, and the removal of the template molecule by extraction with an organic solvent results in the appearance of molecular imprints in the polymer, complementary to the template molecule.
Sorption and regeneration of a sensor layer based on nanoparticles of ractopamine molecularly imprinted polymer by piezoelectric quartz weighing. MIP MA–EGDMA nanoparticles in a solution of cyanoacrylate in tetrahydrofuran were immobilized on the surface of the sensor gold electrode until template molecules were removed from them. For the repeated use of the sensor for the determination of ractopamine, it is necessary to choose a composition of the regenerating solution, which removes the template from the polymer matrix to the maximum extent with a high rate and does not have a destructive effect on the molecular imprint configuration.
We found experimentally the duration of removal to the maximum extent and of the incorporation of the template into the polymer matrix that ensured reproducible results. It was noted that, in using twice-distilled water (solution 1) and a 25% solution of acetic acid (solution 2) as regenerating solutions, the largest amount of ractopamine was removed in the first 1‒2 min, and then the film begun to swell and bind water. The repeated incorporation of template molecules was not observed, possibly, due to the effect of “rubbing prints,” as well as due to the filling of prints with water molecules. Only in using solution 3 in 16 min, the template was almost completely removed from the surface of MIP particles, so this regenerating solution was chosen for the further studies.
For the subsequent use of the MIP layer to determine ractopamine, it was necessary to establish the duration of the re-incorporation of template molecules into molecular prints. It was found that the maximum amount of ractopamine 0.82 μg was incorporated in 3 min (Fig. 4), and with a further contact of the MIP layer with the ractopamine solution, the mass of the polymer film decreased.

The dependence of the mass of ractopamine embedded in molecular imprints on the duration of contact with an aqueous solution.
The characteristics of the MIP-based piezoelectric sensor (Table 3) indicate a possibility of its use for determining ractopamine in liquid media.
A comparison of the masses of embedded ractopamine during repeated and subsequent sorption shows that they differ slightly (ΔmREM(Ract) = 0.71 ± 0.08 and Δ\(m_{{{\text{REM(Ract)}}}}^{'}\) = 0.69 ± 0.05 μg) and are significantly less than the mass of ractopamine (1.0 ± 0.1 μg) included in the MIP structure during the synthesis. The differences in the concentration of molecular imprints calculated after the repeated and subsequent incorporation of the template molecules R' (2.08 mmol/cm2) and R'' (2.04 mmol/cm2) are also insignificant. Therefore, a constant number of molecular imprints conveniently located for the re-incorporation of template molecules forms only after the second sorption–desorption cycle. At the same time, in the initial regeneration of particle surface, a partial destruction of polymer structures adjacent to the imprint probably occurs, which explains the higher value of DmEM(Ract) = 1.03 μg established by piezoelectric quartz micro-weighing.
The ratio ΔFMIP : ΔFNIP = 6 : 1 makes it possible to compare the contributions of specific and non-specific interactions to the analytical signal of the sensor.
Selectivity of particles of MA–EGDMA ractopamine molecularly imprinted polymer immobilized on the surface of the electrode of the piezoelectric quartz sensor was evaluated using the cross-reactivity coefficient CR, % (Table 4).
The high selectivity of MIP with respect to ractopamine in the presence of clenbuterol and salbutamol, which, like ractopamine, belong to the group of beta-agonists and have similar structures, was revealed. Selectivity in the presence of penicillin and cefotaxime was also high and indicated a possibility of determining ractopamine in the presence of equal and even higher concentrations of antibiotics. If the target and interfering compounds are present in a sample in concentrations of 1 mg/mL or lower, ractopamine can be selectively determined in the presence of beta-agonists (clenbuterol and salbutamol) and beta-lactams (penicillin and cefotaxime). Only at a significant (10-fold) excess of clenbuterol and salbutamol in the analytical sample, they will interfere with the determination of ractopamine.
REFERENCES
Vaihinger D., Landfester, K., Krauter, I., Brunner, H., and Tovar, G.E.M., Macromol. Chem. Phys., 2002, vol. 203, no. 13, p. 1965.
Poma, A., MIPs Nanoparticles, BioNanoTech, 2005.
Poma, A., Turner, A.P.F., and Piletsk, S.A., Trends Biotechnol., 2010, vol. 203, p. 629.
Zhu, H., Wang, Y., Yuan, Y., and Zeng, H., Anal. Methods, 2010, vol. 10, p. 139.
Yoshimatsu, K., Reimhult, K., Krozer, A., Mosbach, K., and Ye, L., Anal. Chim. Acta, 2007, vol. 584, p. 112.
Ye, L. and Haupt, K., Anal. Chem., 2004, vol. 378, p. 1887.
Baggiani, C., Anfossi, L., and Giovannoli, C., Analyst, 2008, vol. 133, p. 719.
Ermolaeva, T.N., Chernyshova, V.N., and Bessonov, O.I., Sorbtsionnye Khromatogr.Protsessy, 2015, vol. 15, no. 3, p. 345.
Avila, M., Zougagh, M., and Rios, A., TrAC,Trends Anal. Chem., 2008, vol. 27, no. 1, p. 54.
Lai, J.-P., Yang, M.-L., Niessner, R., and Knopp, D., Anal. Bioanal. Chem., 2007, vol. 389, no. 2, p. 405.
Li, P., Rong, F., and Yuan, C., Polym. Int., 2003, vol. 52, no. 12, p. 1799.
Xu, Sh., Li, J., and Chen, L., Talanta, 2011, vol. 85, p. 282.
Abouzarzadeh, A., Forouzani, M., Jahanshahi, M., and Bahramifarb, N., J. Mol. Recognit., 2012, vol. 25, p. 404.
Pardeshi, S., Dhodapkar, R., and Kumar, A., Food Chem., 2013, vol. 33, p. 239.
Barahona, F., Turiel, E., Cormack, P.A.G., and Martin-Esteban, A., J. Polym. Sci., Part A: Polym. Chem., 2010, vol. 48, p. 1058.
Barahona, F., Turiel, E., Cormack, P.A.G., and Martin-Esteban, A., J. Sep. Sci., 2011, vol. 34, p. 217.
Pérez, N. and Mayes, A.G., Langmuir, 2004, vol. 20, p. 3775.
Lu, C.H., Wang, Y., Li, Y., Yang, H.H., Chen, X., and Wang, X.R., J. Mater. Chem., 2009, vol. 19, p. 1077.
Arenas, L.F., Ebarvia, B.S., and Sevilla, F.B., Anal. Bioanal. Chem., 2010, vol. 397, p. 3155.
Liu, N., Han, J., Liu, Z., Qu, L., and Gao, Z., Anal. Methods, 2013, vol. 5, p. 4442.
Reimhulta, K., Yoshimatsub, K., Risvedenb, K., Chena, S., Yeb, L., and Krozera, A., Biosens. Bioelectron., 2008, vol. 23, p. 1908.
Ermolaeva, T.N., Farafonova, O.V., and Bessonov, O.I., Sorbtsionnye Khromatogr.Protsessy, 2019, vol. 19, no. 6, p. 682.
Karaseva, N., Ermolaeva, T., and Mizaikoff, B., Sens. Actuators, B, 2016, vol. 225, p. 199.
Ebarvia, B.S., Ubando, I.E., and Sevilla, F.B., Talanta, 2015, vol. 1, p. 1260.
UN Food Safety Body Sets Limits on Veterinary Growth Promoting Drug. Codex Alimentarius Commission Adopts Maximum Residue Levels, 2012. http://www.fao.org/ news/story/en/item/150953/icode. Accessed April 29, 2020.
Residue Valuation of Certain Veterinary Grugs, Meeting 2010: Evaluation of Data on Ractopamine Residues in Pig Tissues, Joint FAO/WHO Expert Committee on Food Additives, Rome, Italy, 2010, p. 52.
Liu, M., Ning, B., Qu, L., Peng, Y., Dong, J., Gao, Na., Liu, L., and Gao, Zh., Sens. Actuators, B, 2012, vol. 161, no. 1, p. 124.
Poo-arporn, Y., Pakapongpan, S., Chanlek, N., and Poo-arporn, R.P., Sens. Actuators, B, 2019, vol. 284, p. 164.
Felix, F.S. and Angnes, L., Biosens. Bioelectron., 2018, vol. 102, p. 470.
Pan, M., Li, R., Xu, L., Yang, J., Cui, X., and Wang, Sh., Sensors, 2018, vol. 18, p. 1870.
Ermolaeva, T.N., Farafonova, O.V., and Bessonov, O.I., J. Anal. Chem., 2019, vol. 74, no. 9, S1.
Yang, K. and Ye, L., Macromolecules, 2009, vol. 42, p. 8739.
Funding
This work was supported by the Russian Foundation for Basic Research, project No. 13-03-97505-r_center_a “Selectivity and efficiency of molecular recognition and determination of hormones and beta-agonists using a piezoelectric quartz immuno- and biomimetic sensor.”
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Kudrinskaya
Rights and permissions
About this article

Cite this article
Ermolaeva, T.N., Farafonova, O.V., Chernyshova, V.N. et al. A Piezoelectric Sensor Based on Nanoparticles of Ractopamine Molecularly Imprinted Polymers. J Anal Chem 75, 1270–1277 (2020). https://doi.org/10.1134/S1061934820100068
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934820100068