
The stability of the atomic structure and physical and chemical properties of new materials is of crucial importance for their practical use. Long-term (about 120 000 min) and regular monitoring of the molecular structure of two samples of poly(vinylidene fluoride) (PVDF) film after its five-hour chemical dehydrofluorination and rinsing with ethanol are performed using infrared (IR) spectroscopy. The samples differed in low-pressure exposure duration before measurement. Changes in the absorption spectra of the stretching vibrations of triple carbon-carbon bonds are analyzed in the wave-number range of 2000–2300 cm–1. The initial PVDF film is transparent in this region, but, after dehydrofluorination, an absorption band of complex shape is observed in it. Visually, the band consists of three wide features with centers around 2050, 2100, and 2160 cm–1. The first and the second bands are absent immediately after synthesis, but, as the samples age, they appear and grow proportionally to each other and time intervals of rapid and slow growth, as well as a stabilization period, are observed.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
In recent decades, there has been an increasing number of papers devoted to the synthesis and study of new materials based on carbon (graphene, carbon nanotubes, and fullerenes). Interest in carbon materials is due to their unique physical and chemical properties which are promising for practical application. The synthesis and research of low-dimensional carbon structures containing chain fragments [1–10] is still an urgent scientific problem. In this regard, the hypothesis of the possibility of a one-dimensional modification of chemically pure carbon (carbyne) [1] attracts attention. It is assumed that ideal carbyne consists of linear carbon chains in which neighboring atoms are connected by either double or alternating triple and single bonds (cumulene or polyyne ordering types, respectively). A number of studies have calculated the width of the carbyne band gap and various calculation methods have led to a wide variety of values in the range from 0.2 to 8.5 eV [11]. At the same time, most experimental studies show that carbyne-like structures have semiconductor properties [1, 12, 13]. There are various and often contradictory structural models of carbyne chains and their mutual ordering [1, 2, 4]. The discovery of crystalline carbyne in natural minerals [3] shows the fundamental possibility of its laboratory synthesis and, as a result, the prospect of its practical application.
However, perfect carbyne crystals have not been synthesized yet. The synthesis products, called “carbynoids”, contain not only nanoscale fragments of linearly polymerized carbon, but also a large number of defects (non-carbon inclusions, inter-chain crosslinking, etc.) [1, 2, 4]. This type of carbon was first synthesized in the USSR in the 1960s [1].
One of the ways to synthesize carbyne-like nanostructures is to carbonize chain polymers that have a carbon base, such as poly(vinylidene fluoride) (PVDF). PVDF itself has a number of useful properties, as a result of which it is widely used in membrane technologies [14], electronics, medicine, acoustics, etc. [15, 16]. Its molecules are carbon chains, to each atom of which two atoms of fluorine and hydrogen are alternately attached. There are three main types of chain conformation: α, β, and γ [17]. The polymer material can have different properties depending on the prevailing conformational type. For example, when β-type chains (plane zigzag) dominate, the polymer has piezoelectric properties. As a rule, crystalline and amorphous regions coexist in PVDF.
Two main ways of PVDF carbonation are known: radiative (irradiation with photons and bombardment with microparticles of different mass and energy) [18–27] and chemical [1, 28–34] ones. PVDF contains the equal number of fluorine and hydrogen atoms, which are removed from the polymer in the form of hydrogen fluoride as a result of radiation or chemical exposure. This allows deep, though not complete [26], carbonization of PVDF without destroying the carbon-chain structure. Neighboring carbon atoms, freed from fluorine and hydrogen, can be connected to each other by single and multiple bonds, forming cumulene or polyyne structures [1]. In this case, inter-chain crosslinking also become possible. However, incomplete carbonization results in the retention of a certain number of CF2 (CH2) and/or CF (CH) molecular complexes within a partially “naked” chain. These complexes can play a stabilizing role by keeping carbonized fragments of neighboring chains at a distance sufficient to prevent the immediate collapse of linear carbon into higher-dimensional structures.
The simplest and most productive method of deep PVDF carbonization, which allows the modification of a rather large amount of polymer, is chemical processing. According to the well-known model of chemical carbonization, fluorine and hydrogen are separated from the carbon skeleton in equal amounts under the influence of a liquid dehydrofluorinating mixture [1]. To date, some characteristic features of chemically synthesized PVDF derivatives have been identified [34]. Estimates of the depth of penetration of the dehydrofluorinating mixture into the partially crystalline PVDF film were made at various times of dehydrofluorination. The conductivity of the carbonized layer was higher than that of its polymer precursor [35], which has the prospect of synthesizing conductive and/or semiconductor nanofilms on an elastic and transparent dielectric substrate for optoelectronic and acoustoelectronic nanodevices.
The study of PVDF chemical carbonization products by nuclear magnetic resonance (NMR) and IR spectroscopy showed that an increase in the duration of treatment in a dehydrofluorinating mixture increases the ratio of the number of protons to the number of fluorine nuclei, despite a synchronous and steady decrease in the number of CH2 and CF2 groups [34]. This is due to the intense reaction of the nucleophilic substitution of fluorine atoms with hydroxyl groups contained in the components of the mixture and atmospheric air. This effect prevents the formation of multiple carbon-carbon bonds, inherent in carbynoid structures.
In [36], we were able to reduce the addition of OH groups to the carbon base by replacing distilled water with ethanol during the final washing of a sample that was chemically carbonized for 30 min. The evolution of its molecular structure during aging was regularly monitored for almost an entire calendar year by IR spectroscopy. A long-term non-monotonic change in the shape of the IR spectra in the frequency range corresponding to the absorption band of triple carbon-carbon bonds was detected. It is this absorption band that is of particular interest, since the existence of triple carbon-carbon bonds is possible only in the chain (carbynoid) form of carbon.
This paper describes the results of modification of this IR absorption band during the aging of a carbynoid film synthesized by the chemical dehydrofluorination of PVDF for five hours. The tenfold increase in the reaction duration compared to [36] has two goals: to increase the concentration of triple carbon-carbon bonds because of a larger amount of the reacted polymer substance and (possibly) to reduce the time elapsed after sample synthesis after which its molecular structure will no longer change. It is obvious that stability of the atomic structure and physical and chemical properties of new materials, as well as identification of the necessary conditions for their storage and operation, are of key importance for their practical use. It was shown in [37] that the kinetics of the deactivation of paramagnetic centers in chemically carbonized PVDF samples is significantly influenced by interaction with air. In this regard, we further study the influence of atmospheric air on the nature and kinetics of modification of the molecular structure of a chemically carbonized material and obtain a completely unexpected result.
OBJECT AND RESEARCH METHODOLOGY
The material under study was obtained by the five-hour dehydrofluorination of a 20-mm thick PVDF F-2m film produced by Plastpolymer (Saint Petersburg) at room temperature. The dehydrofluorinating mixture consisted of one volume part of a saturated solution of caustic potassium in ethanol and nine volume parts of chemically pure acetone. The synthesis procedure is almost the same as that described in detail in [34], except that after pre-washing with acetone, final washing of the sample was performed with ethanol instead of water [36]. As a result of chemical exposure, the initially transparent polymer film became completely matte black. To study the effect of atmospheric air, the dehydrofluorinated film was divided into two parts immediately after the end of synthesis and washing. Both parts were strictly fixed in separate holders with rectangular windows. This made it possible to ensure the identical passage of IR radiation through the same areas of the samples during subsequent measurements. Before the measurements, both samples were kept in a pre-vacuum chamber at room temperature and a residual gas pressure of about 5 × 10–2 mm Hg for 1 and 13 days (samples 1 and 2, respectively). We believed that a reduced pressure could inhibit the aging processes of sample 2 at the initial stage and expected to observe a corresponding delay in the course of expected modification of its molecular structure compared to sample 1. The samples were stored in the same chamber between measurements, specifically at night.
As a rule, the use of only one experimental method cannot provide exhaustive information about the object under study. However, the IR spectroscopy technique is highly sensitive to the minor modification of the molecular structure of a substance [38]. Thus, it was this method that was chosen by us to monitor the aging process of the synthesized material. The IR spectra were measured in the frequency range of 400–4000 cm–1 using a Shimadzu IRAffinity-1 Fourier spectrometer in the transmission geometry. The measurements started at 1500 and 18 406 minutes and ended at 116 528 and 116 530 minutes (the samples 1 and 2, respectively) after chemical treatment and washing. A total of 230 and 171 spectra of samples 1 and 2 were obtained. This allowed us to observe in detail the change in the shape of the IR spectra during aging. The experimental data on transmission were recalculated by the optical density.
EXPERIMENTAL RESULTS AND DISCUSSION
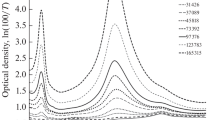
The most significant irreversible modification of the shape of the IR spectra at an increase in the duration of aging of both samples is observed in the frequency range of 1800–2300 cm–1. The original PVDF film is transparent in this region, but, after dehydrofluorination, there is absorption in the form of a band of complex shape. It is known [36, 38] that the 2000–2300 cm–1 absorption band arises as a result of the stretching vibrations of triple carbon-carbon bonds. Figures 1 and 2 show corresponding fragments of the IR spectra of samples 1 and 2 measured at various points in time after the end of synthesis. The shape of the spectra during aging of the samples changes exclusively in a regular way. Basically, modification occurs because of the appearance and growth of fairly wide absorption peaks near 2050 and 2100 cm–1 as the duration of aging increases.

Modification of the shape of the absorption band of triple carbon-carbon bonds during aging of sample 1. Recording of the spectra presented as an example was performed 2942, 22 893, 34 646, 50 342, 50 488, 61 892, 114 997, and 116 725 minutes after the end of synthesis. The growth of absorption peaks near 2050 and 2100 cm–1 occurs as the duration of aging increases, so the curves in the figure are easily identified.

Modification of the shape of the absorption band of triple carbon-carbon bonds during aging of sample 2. Recording of the spectra presented as an example was carried out 24 520, 30 009, 34 645, 50 489, 61 856, 83 466, 102 221, and 116 727 minutes after the end of synthesis. The growth of absorption peaks near 2050 and 2100 cm–1 occurs as the duration of aging increases, so the curves in the figure are easily identified.
From the comparison of Figs. 1 and 2, we can clearly see the qualitative similarity of modification of the shape of the spectra of both samples. If you look closely at the figures, you can also notice differences. It can be seen that absorption near 2050 and 2100 cm–1 in the spectra of sample 2 is less than that of sample 1 if the spectra of both samples are recorded at close points in time. To do this, it is enough to compare, e.g., the third spectra at the bottom in the two figures, which were recorded almost simultaneously (34 646 and 34 645 minutes), or the first ones at the top (116 725 and 116 727 minutes).
To parameterize the shape of the spectra, the background component, which was approximated by a straight line segment in the range of wave numbers 1830–2240 cm–1, was first uniformly subtracted from them and then the optical density values were measured at the following points: 2050, 2102, and 2162 cm–1. These points correspond to the frequency positions of the maxima of three clearly observed peaks, which we believe are related to the valence vibrations of C≡C-bonds. As a result of a change in the shape of the spectra, some peaks turn into shoulders and vice versa; therefore, the maxima of all three peaks cannot be observed explicitly at the same time points. Further, we agree to coin these three peaks or shoulders as spectral features 1–3 with centers near 2050, 2102, and 2162 cm–1. Another broad spectral feature in the region of 1850–2000 cm–1 occurs most likely as a result of stretching vibrations of cumulative C=C-bonds of various lengths [36] and its shape changes as a result of the partial superposition with the low-frequency shoulder of feature 1.
Figure 3 shows the dependences of the optical density values at the centers of features 1–3 on the duration of aging of the sample 1. This dependence for sample 2 is qualitatively very similar, so we did not give it in a separate figure. Feature 3 changes slightly in the spectra of both samples. The non-monotonic synchronous changes in features 1 and 2 are most noticeable. This connection can be clearly seen in Figs. 1–3. We constructed the corresponding trend lines using Excel software and made sure that the dependences between the optical density values D1 and D2 at the centers of features 1 and 2 for both samples are very well described by linear equations with fairly close coefficients:

Dependence of the optical density of features (⚫) 1, (◻) 2, and (×) 3 on the duration of aging of the sample 1.
D1 = 2.34D2 – 0.15 (the correlation coefficient R2 = 0.997, sample 1) and
D1 = 2.37D2 – 0.17 (the correlation coefficient R2 = 0.997, sample 2).
This obvious relationship indicates the influence of the same molecular process that occurs during aging of the samples on the growth of both features.
The rate of increase in IR absorption in the range of 2000–2250 cm–1 differs significantly at different aging stages. Both features under discussion show weak growth up to about 49 000 minutes after the end of synthesis and then grow very quickly up to 56 200 minutes. In the range of 56 200–74 000 min, slow growth is again observed followed by stabilization of the optical density up to a new increase after 100 000 min. The described non-monotonic increase in the absorption is well illustrated in Fig. 4, which shows variations in the value of the optical density D1 at the center of feature 1, dominant in the spectra depending on the aging duration of both samples. Figure 4 also clearly shows that, (as noted above) at close moments of time, absorption at about 2050 cm–1 in the spectra of sample 2 is less than that of sample 1. The synchronous changes in parameter D1 (as well as those of parameter D2, which is not shown in Fig. 4) in the spectra of both samples up to 100 000 min, including the time interval of rapid growth of these parameters, seem unexpected and yet inexplicable to us. This synchronicity is violated at above 100 000 min: a new jump in the parameters D1 and D2 in the spectra of sample 2 occurs 21 758 min earlier, compared to that of sample 1.

Nonmonotonic changes in the optical density D1 as the samples 1 (⚫) and 2 (◻) age.
We note another constantly occurring reversible effect, the cause of which is still unclear. Figures 3 and 4 show a series of nearly vertically arranged experimental points. These are the values of the parameters D1 and D2 in the spectra obtained at close moments of time (after 20–30 min) in a single series of measurements. As noted above, the samples were stored at a reduced pressure between the measurements. When recording the first spectrum of the sample just extracted from the vacuum chamber, the parameters D1 and D2 have as a rule the highest value in this series of measurements and then gradually decrease.
This effect is demonstrated in Figs. 5 and 6 by the example of two IR spectra of sample 1 recorded at 87 637 and 87 957 minutes of aging at the beginning and end of one series of measurements. The first was obtained immediately after extraction from the vacuum chamber and the second, after 320 min of being exposed to air. Presumably, the observed phenomenon can be related to the gradual accumulation of moisture on the surface of the samples over the series of measurements. This is indicated by an increase in absorption in the spectral region of 2700–3700 cm–1, which is typical for the stretching oscillations of OH groups (Fig. 6). This means that the surface of the samples becomes hygroscopic as a result of dehydrofluorination. It is likely that increasing the water layer thickness during successive measurements in air leads to a gradual decrease in the intensity of radiation passing through the sample. It is also possible that the small peaks observed in all spectra at 653, 680, and 850 cm–1 are the result of the libration fluctuations of water clusters of various sizes [39, 40]. Some combination modes of the libration and bending vibrations of such clusters can contribute to the absorption band characteristic of stretching vibrations of C≡C-bonds [40].

Fragments of the IR spectra of sample 1 in the range of 1800–2250 cm–1, measured immediately after extraction from the vacuum chamber at 87637 minutes of aging (solid line) and after 320 minutes of exposure to air (dotted line). Previously, the linear component of the background was subtracted from the spectra.

Fragments of the IR spectra of sample 1 in the range of 2600–3800 cm–1, measured immediately after extraction from the vacuum chamber at 87637 minutes of aging (solid line) and after 320 minutes of exposure to air (dotted line).
In all the spectra obtained in this study, a CH band of IR absorption is observed against the background of the OH band. It is a superposition of the absorption bands, which in terms of their frequency positions are very close to those typical for four types of valence vibrations of molecular complexes in ethanol: symmetric and asymmetric vibrations of CH groups at frequencies of 2870 (sСН2), 2894 (sСН3), 2927 (aСН2), and 2975 (aСН3) cm–1, respectively [41, 42]. However, in general, the form of the CH and OH bands differs from that of ethanol. By analogy with [36], we can assume the presence of ethoxy groups attached to the fragments of carbon chains freed from fluorine in the carbonized material. However, both the CH-band shape and intensity were exceptionally stable in this study in contrast to the results in [36]. In particular, the maximum growth rate of the D1 and D2 parameters is observed between 48 920 and 56 200 minutes of aging for both samples. During this time, the area of the absorption band in the range of 1830–2240 cm–1 increases by 1.7 and 2.1 times and the area of the CH band decreases only by 1.6 and 7% for samples 1 and 2, respectively. This can indirectly indicate that a decrease in the number of ethoxy groups is not the cause of an increase in absorption because of the contribution of C≡C-bond vibrations. The observed changes can be the result of either a real increase in the concentration of triple bonds in the sample or changes in the symmetry of their environment by various functional groups, in particular, water clusters.
In contrast to the ethanol spectrum, there is also a feature with a center near 3090–3100 cm–1 (Fig. 6), which is present in the spectra at all stages of sample aging. Its occurrence during dehydrofluorination of the Kynar PVDF film was observed by us earlier [34]. Unambiguous identification of the origin of this feature is currently also not possible and requires further study.
CONCLUSIONS
Our long-term experiment on the aging of chemically dehydrofluorinated PVDF raised more questions than it answered. To date, as a result of the measurements, it has only been possible to identify some features of the phenomenon under study.
(1) After the end of chemical synthesis, the sample molecular structure changes over a long time. These changes are appropriately described by the term “aging”.
(2) In the frequency range typical for the manifestation of stretching vibrations of triple bonds, there is a strong change in the shape of the IR absorption spectra, indicated in the appearance and growth of two broad features with centers at about 2050 and 2100 cm–1. Their evolution is surprisingly synchronous and proportional, which is an indirect sign of reflection in it of the same process in modification of the molecular structure of the sample. An alternative explanation for this relationship could be based on the assumption that feature 2 itself does not change, but the growth of feature 1 causes its proportional increase relative to the background line. However, this explanation contradicts experimental facts, since feature 2 is not observed in the spectra even at the earliest stages of aging, when feature 1 is either absent or very weak.
(3) Analysis of the IR spectra did not allow us to clearly determine the reason for the increase in absorption in the frequency range characteristic of the stretching vibrations of C≡C-bonds. The observed changes can be the result of either an actual increase in the concentration of triple bonds in the sample or changes in the symmetry of their environment by different functional groups.
(4) A dependence of the evolution of the molecular structure during aging on the duration of the initial exposure of the dehydrofluorinated samples at low pressure was not explicitly found. In the spectra of both samples, there is a synchronous powerful increase in the contribution of the stretching vibrations of C≡C bonds in the time range of 49 000–56 200 min after synthesis.
(5) Significant hygroscopicity of the sample under study, which is manifested in a change in the spectrum in the region of OH-bond vibrations, was detected. When the sample is removed from the vacuum chamber, the intensity of the OH band is minimal at the very beginning of the next series of measurements and features 1 and 2, on the contrary, are the most intense. In each subsequent measurement, there is first a gradual increase in the absorption of OH bonds and then the achievement of some limiting value for the given series of measurements. In this case, features 1 and 2, although slightly, noticeably and synchronously decrease. It is possible that an increase in the thickness of the water layer during successive measurements in air leads to a gradual decrease in the intensity of the radiation passing through the sample. The presence of water in the samples does not exclude its influence on the shape of the spectra also in the range of 2000–2200 cm–1.
REFERENCES
Carbyne and Carbynoid Structures, Ed. by R. B. Heimann (Kluwer Acad., Dordrecht, 1999).
W. Luo and W. Windl, Carbon 47, 367 (2009). https://doi.org/10.1016/j.carbon.2008.10.017
T. G. Shumilova, Yu. V. Danilova, M. V. Gorbunov, et al., Dokl. Akad. Nauk. 436, 394 (2011).
E. A. Belenkov and I. V. Shakhova, Phys. Solid State 53, 2385 (2011).
A. Freitas, S. Azevedo, and J. R. Kaschny, Physica E 84 444 (2016). https://doi.org/10.1016/j.physe.2016.07.018
Y. Prazdnikov, Journal of Modern Physics. 3 (9), 895 (2012). https://doi.org/10.4236/jmp.2012.39117
E. A. Buntov, A. F. Zatsepin, M. B. Guseva, et al., Carbon 117, 271 (2017). https://doi.org/10.1016/j.carbon.2017.03.010
C.-S. Kang, K. Fujisawa, Y.-I. Ko, H. Muramatsu, et al., Carbon 107, 217 (2016). https://doi.org/10.1016/j.carbon.2016.05.069
N. F. Andrade, T. L. Vasconcelos, C. P. Gouvea, et al., Carbon 90, 172 (2015). https://doi.org/10.1016/j.carbon.2015.04.001
D. A. Kovriguine and S. P. Nikitenkova, Phys. Solid State 58, 611 (2016).
L. Ravagnan, F. Siviero, C. Lenardi, et al., Phys. Rev. Lett. 89, 285506 (2002). https://doi.org/10.1103/PhysRevLett.89.285506
L. Shi, P. Rohringer, M. Wanko, et al., Phys. Rev. Mater. 1 (7), 075601 (2017). doi . Rev. Materials.1. 075601https://doi.org/10.1103/Phys
V. V. Korshak, Yu. P. Kudryavtsev, V. V. Khvostov, et al., Carbon 25 (6), 735 (1987). https://doi.org/10.1016/0008-6223(87)90143-6
S. E. Evsyukov and V. G. Babaev, et al., Carbon 30 (2), 213 (1992). https://doi.org/10.1016/0008-6223(92)90082-8
S. Zhang, J. Shen, X. Qiu, et al., J. Power Sources 153, 234 (2006). https://doi.org/10.1016/j.jpowsour.2005.05.020
A. Kimoto and N. Sugitani, Meas. Sci. Technol. Medical and Healthcare Textiles, Ed. by S. C. Anand (Wood Head, Manchester, 2010), 21, 075202 (2010). https://doi.org/10.1088/0957-0233/21/7/16
V. V. Kochervinskii, Usp. Khim. 65, 936 (1996).
A. Le Moel, J. P. Duraud, and E. Balanzat, Nucl. Instrum. Methods Phys. Res., Sect. B 18 (1–6), 59 (1986). https://doi.org/10.1016/S0168-583X(86)80012-X
E. H. Adem, S. J. Bean, C. M. Demanet, et al., Nucl. Instrum. Methods Phys. Res., Sect. B 32 (1–4), 182 (1988). https://doi.org/10.1016/0168-583X(88)90206-
M. D. Duca, C. L. Plosceanu, and T. Pop, J. Appl. Polym. Sci. 67 (13), 2125 (1998). https://doi.org/10.1002/(SICI)1097-4628(19980328)67:13<2125::AID-APP2>3.0.CO;2-
M. M. Brzhezinskaya, V. M. Morilova, E. M. Baitinger, et al., Polym. Degrad. Stab. 99, 176 (2014). https://doi.org/10.1016/j.polymdegradstab.2013.11
S. S. Chebotaryov, A. A. Volegov, L. A. Pesin, et al., Physica E 36 (2), 184 (2007). https://doi.org/10.1016/j.physe.2006.10.011
S. S. Chebotaryov, E. M. Baitinger, A. A. Volegov, et al., Radiat. Phys. Chem. 75 (11), 2024 (2006). https://doi.org/10.1016/j.radphyschem.2005.12
L. A. Pesin, V. M. Morilova, D. A. Zherebtsov, et al., Polym. Degrad. Stab. 98 (2), 666 (2013). https://doi.org/10.1016/j.polymdegradstab.2012.11
A. Le Moel, J. P. Duraud, C. Lecomte, et al., Nucl. Instrum. Methods Phys. Res., Sect. B 32 (1–4), 115 (1988). https://doi.org/10.1016/0168-583X(88)90192-926
A. L. Sidelnikova, V. P. Andreichuk, L. A. Pesin, et al., Polym. Degrad. Stab. 110, 308 (2014). https://doi.org/10.1016/j.polymdegradstab.2014.09.009
E. Morikawa, J. Choi, H. M. Manohara, et al., J. Appl. Phys. 87 (8), 4010 (2000). https://doi.org/10.1063/1.372447
G. J. Ross, J. F. Watts, M. P. Hill, et al., Polymer 41, 1685 (2000). https://doi.org/10.1016/S0032-3861(99)00343-2
G. J. Ross, J. F. Watts, M. P. Hill, and P. Morrissey, Polymer 42, 403 (2001). https://doi.org/10.1016/S0032-3861(00)00328-1
Yu. P. Kudryavtsev, S. E. Evsyukov, and V. G. Babaev, Russ. Chem. Bull. 41, 966 (1992).
I. G. Margamov, L. A. Pesin, Yu. P. Kudryavtsev, and S. E. Evsyukov, Appl. Surf. Sci. 148 (3–4), 183 (1999). https://doi.org/10.1016/S0169-4332(99)00154
N. A. Mavrinskaya, L. A. Pesin, M. Baumgarten, et al., Magn. Reson. Solids EJ 10 (1), 31 (2008).
V. E. Zhivulin, N. A. Moskvina, I. V. Gribov, et al., J. Surf. Invest.: X-ray, Synchrotron Neutron Tech. 11, 927 (2017).
V. E. Zhivulin, V. M. Chernov, A. A. Osipov, et al., Phys. Solid State 59, 1414 (2017).
L. A. Pesin, S. S. Chebotaryov, A. M. Kuvshinov, et al., J. Surf. Invest.: X-ray, Synchrotron Neutron Tech. 4, 214 (2010).
V. E. Zhivulin, L. A. Pesin, E. A. Belenkov, V. A. Greshnyakov, N. A. Zlobina, and M. M. Brzhezinskaya, Polym. Degrad. Stab 172, 109059 (2020).
V. E. Zhivulin, N. A. Zlobina, and L. A. Pesin, Bull. Tomsk Polytech. Univ. Geo Assets Engineering 326 (10), 150 (2015).
N. L. Alpert, W. E. Keiser, and H. A. Szymanski, IR: Theory and Practice of Infrared Spectroscopy (Springer, 2012).
V. Pogorelov, I. Doroshenko, G. Pitsevich, et al., J. Mol. Liq. 235, 7 (2017). https://doi.org/10.1016/j.molliq.2016.12.037
A. Vasylieva, I. Doroshenko, Ye. Vaskivskyi, et al., J. Mol. Struct. 1167, 232 (2018). https://doi.org/10.1016/j.molstruc.2018.05.002
G. A. Pitsevich, I. Yu. Doroshenko, V. E. Pogorelov, et al., Am. J. Chem. 2 (4), 218 (2012). https://doi.org/10.5923/j.chemistry.20120204
G. A. Pitsevich, I. Yu. Doroshenko, V. E. Pogorelov, et al., Fiz. Nizk. Temp. (Kiev) 39 (4), 499 (2013).
ACKNOWLEDGMENTS
We thank Prof. I. Y. Doroshenko for her interest in the work, useful discussion of the experimental results, and valuable comments on their interpretation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by N. Petrov
Rights and permissions
About this article

Cite this article
Zhivulin, V.E., Khairanov, R.K., Zlobina, N.A. et al. Modification of the IR Spectra Shape in the 2000–2300 cm–1 Absorption Band upon the Aging of a Chemically Dehydrofluorinated Poly(vinylidene fluoride) Film. J. Surf. Investig. 14, 1144–1151 (2020). https://doi.org/10.1134/S1027451020060178
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1027451020060178