
Abstract
TiO2 films with a 1D nanotube structure were obtained by electrochemical anodic oxidation of titanium foil. Electrochemical reductive activation of the TiO2-nanotube-based electrodes was carried out using the method of cyclic voltammetry. The activated electrodes showed significantly higher current density and quantum efficiency of the photoelectrochemical water splitting as compared to native TiO2 nanotubes. Electrochemical treatment of the electrodes by the cyclic voltammetry leads to increase in the photocurrent density by a factor of 4 to 14, depending both on the used wavelength and applied potential. The analysis of electrochemical impedance spectra showed that the increase in the photoelectrochemical process performance is due to increase in the charge transfer rate at the semiconductor/electrolyte interface, as well as improved electronic conductivity of the oxide layer, which contributes to better charge carrier separation and decrease in their recombination rate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Photocatalytic properties of TiO2-based systems stipulated their wide application in the development of processes of organic pollutant degradation, production of environmentally-friendly fuels, water photo-splitting, as well as anodes of solar cells and sensors [1–8].
TiO2-nanotubes obtained by electrochemical anodizing of titanium foil is an effective structure for the photocatalytic water splitting. Large surface area and unidimensional architecture of the TiO2-nanotubes oriented vertically with respect to substrate is favorable for the creation of the electrode/electrolyte developed surface and accelerating of the transfer of photogenerated charge carrier. Nonetheless, there exist two principal drawbacks preventing their application as an effective photoanode for the water decomposition: low activity in the visible part of solar spectrum, because of large gap (3.2 eV for the anatase) and high recombination rate of electron–hole pairs [4].
It is known that reduced TiO2 (TiO2 – х) contains Ti3+ and oxygen vacancies that create localized states in the gap. The introducing of vacant states Ti3+ or oxygen improved the absorbability and electron conductivity and as a consequence of this increased the photocatalytic activity of the nanostructured-TiO2-based electrodes [8].
To prepare О2-deficienced titanium dioxide nanostructures, different titanium dioxide reduction methods are used. They include: high-temperature hydration, reduction by metals (Al, Zn, Mg) [9], solvo- and ionothermal methods [10, 11], plasma treatment [12], and laser treatment [13]. However, these methods require rather severe conditions or expensive equipment. On this reason, the electrochemical reduction at the hydrogen evolution potentials is an alternative for the preparation of titanium dioxide possessing the corresponding localized states. The electrochemical doping with Ti3+ used to be carried out in potentiostatic condition at different cathodic potentials, the process duration being varied [5, 14].
Cyclic voltammetry finds wide application in the studying of the electrochemical reaction kinetics and mechanism, in the studies of semiconductors, supercapacitors, solar cells, and fuel cells [15–18].
In this work, we suggested a new approach to the using of the cyclic voltammetry as a method of improving the activity of nanostructured anodic TiO2 films in the photoelectrochemical water decomposition. We present comparative results of our studies in the photoelectrochemical and electrical characteristics of TiO2-anodes activated by cyclic voltammetry in different electrolytes.
EXPERIMENTAL
Titanium foil (99.9%, 80 μm thick) was subjected successively to ultrasonic cleaning in acetone, alcohol, and deionized water. The cleaned foil was polished chemically by a mixture of reagent grade acids HF : HNO3 : H2O (1 : 1 : 4 v/v), washed by distilled water, and once again cleaned ultrasonically in deionized water, to fully remove possible residues of the acid mixture. The TiO2-nanotube films were obtained by anodizing at the voltage of 50 V for 3 h in the ethylene glycol electrolyte containing 0.3 wt % NH4F (ultra-high purity grade), 98 vol % ethylene glycol (reagent grade), 2 vol % deionized water (resistivity 18.2 MΩ). The electrochemical anodization was performed at standard conditions. Upon the anodizing, the obtained films were dried in air. Then the films were baked in a muffle furnace at a temperature of 450°С for 3 h at a heating rate of 3°С min–1, to convert amorphous TiO2 to the anatase phase.
The electrochemical reductive doping was carried out using the cyclic voltammetry method in 1 М KOH or 0.5 М Na2SO4 aqueous solutions. The process was carried out in a three-electrode cell, using the obtained TiO2-nanotubes-films as a working electrode and platinum helix as auxiliary one. The potential was cycled over the potential range from –1.5 to 2 V vs. Ag/AgCl (sat.) (Е0 = 0.197 V) reference electrode at a potential scanning rate of 100 mV s–1 during 30 cycles.
The structure–morphology characteristics of the titanium dioxide films were obtained by analyzing their images taken by a Hitachi S5500с FEG (FESEM) scanning electron microscope. X-ray diffraction patterns of the samples were taken at a sweeping rate of 1 deg min–1 using a DRON-3 diffractometer (Burevestnik, Russia) with monochromatic CuKα-radiation. In all measurements, the viewing angle was 0.02°.
A FRA BIOLOGIC VSP-300 potentiostat/galvanostat (Bio-LogicSAS, Clarix, France) was used in the evaluating of the samples’ photoelectrocatalytic activity and electrochemical properties. The experiments were carried out on a cooled reactor using a tree-electrode scheme of connection. A Nafion 212 cation-exchange membrane separated the cathodic and anodic compartments. The anolyte was 1 М КОН solution; the catholyte, 0.5 М H2SO4 solution. The anode and cathode were the titanium foil with the synthesized nanotubes (the visible surface area, 2 сm2) and the platinum helix, respectively. The reference electrode was Ag/AgCl (sat.). To avoid the effect of the dissolved oxygen, prior to each experiment both chambers of the reactor were bubbled-through with argon for 30 min. The photocurrent was measured upon UV-A illumination (λ = 400 nm, 90 mW cm–2) and visible light (λ = 450 nm, 50 mW cm–2). The electrochemical impedance was measured at the potential of 0.2 V (vs. Ag/AgCl-electrode) over the frequency range from 100 kHz to 10 mHz with the amplitude of 50 mV.
The amount of evolved hydrogen was estimated volumetrically. The experiments were carried out at the potential of 0.2 V (Ag/AgCl) for 4 h; the evolved hydrogen volume was measured every 30 min.
RESULTS AND DISCUSSION
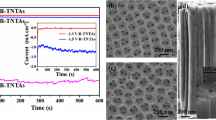
The TiO2 layer prepared by the anodic oxidation is a closely packed highly ordered array of vertically oriented nanotubes (Fig. 1а). The layer thickness determined from the microphotograph is ca. 15 μm; the tube average inner diameter, ca. 100 nm; the tube wall thickness, 10 nm. In Fig. 1b we give X-ray diffraction patterns of the backed nanotubes-layer after the electrochemical activation; it evidenced the titanium dioxide being in the anatase form (JCPDS, 21-1272). The basic peaks of the X-ray diffraction spectrum for the anatase showed no changes; this points that upon the electroreduction the TiO2-nanotubes-films keep their initial phase and crystal structure.

FESEM-image of the obtained nanotubular TiO2-film (а). X-ray diffraction patterns of electrochemically activated TiO2-nanotubes-film (b).
The electrochemical behavior of the water-splitting-photocatalysts was studied prior to and after the activation process by the potential linear sweeping from –1.0 to 1.5 V at the potential scanning rate 10 mV s–1. In Fig. 2а we show current–potential curves taken in dark (curves 4 and 5) and upon UV-A-illumination (curves 1–3) prior to and after the electrochemical doping. The background current of activated electrodes is approximately 8 times as high as that of the initial sample (8 and 1 μА сm–2, respectively, at Е = 0.2 V (Ag/AgCl)); nonetheless, they are negligibly small and increased only at potentials exceeding 1.0 V (Ag/AgCl).

Current–potential curves for titanium dioxide nanofilms: non-activated (1), activated in 1 М КОН (2) and 0.5 М Na2SO4 (3) upon illumination with light with the wavelength λ: 400 (а), 450 nm (b).
Upon the anodic potential shift, electrons trapped in the surface states (Ti–OH) are extracted via assessable states of the conduction band into the external circuit; holes move toward the surface and react with the electrolyte solution and thus contribute to the photocurrent. The maximal photocurrent density (the saturation photocurrent) can be reached at the potential when all surface states have been exhausted. Further positive potential shift has practically no effect on the electron–hole pair recombination rate in the photoanode exhaustion zone [19]. Upon the UV-A-irradiation of the electrochemically activated electrodes, the oxidation photocurrent increased drastically; at that, a much higher anodic potential is required for the reaching of the saturation current, that is, the surface states’ full exhaustion. In particular, no saturation current has been observed at potentials more positive than 2 V (RHE) for the electrode activated in Na2SO4, which points to the high surface state concentration. The photocurrent density increased by a factor of 6–7 as compared with the initial sample at the LED wavelength of 400 nm; by a factor of 4–5, at λ = 450 nm. For convenience in the analysis of the studied electrodes’ photoactivity, the photocurrent densities are summarized in Table 1.
We calculated the quantum efficiency (η) of the photochemical water splitting from the linear curves by the following formula [20]:
where Ip is the photocurrent density, mА сm–2; \(V_{{{\text{rev}}}}^{0}\) is the reversible potential of the standard state for the water splitting reaction, Е = 1.25 V; Vmeas is the applied potential, V; Vocp is the open-circuit potential, V; Pt is the light beam specific power, mW сm–2.
In Figs. 3а, 3b we give the obtained values of η as a function of the applied potential.

Quantum efficiency of electrodes: non-activated (1), activated in 1 М КОН (2) and 0.5 М Na2SO4 (3), upon illumination with light with the wavelength λ: 400 nm (а), 450 nm (b).
The maximal quantum efficiency (ηmax) of the activated electrodes is more than 2–4 times higher than that of the initial TiO2-nanotubes; however, upon the UV-A-irradiation treatment, the potential of ηmax has been shifted to the more positive values. When using a LED with λ = 450 nm for the electrodes activated in Na2SO4, the ηmax value was reached at a potential of ‒0.63 V, which is by 200 mV more negative than in the case of the initial electrode or that activated in КОН. The increase in the quantum efficiency of the activated electrodes can be connected with the increase in the number of surface states and oxygen vacancies involved in the electron trapping and their transmitting into external circuit. On the other hand, the excess oxygen vacancies can serve as recombination centers for the photogenerated charge carriers [21].
To further study the nanostructured TiO2-electrode activity in the water photoelectrochemical splitting, we measured the amount of hydrogen evolved in the reaction. To assure the reliance and confidence of the obtained results, we repeated the measurements 3 times for each electrode. It is to be noted that the amount of evolved Н2 was determined only upon illuminating from a LED with the wavelength of 400 nm because the data concerning the hydrogen evolution at λ = 450 nm could not be correctly interpreted because of large inaccuracy caused by the adopted photoreactor configuration. In Fig. 4 we present kinetics of hydrogen evolution in the reaction of the water photochemical splitting at the TiO2-anodes.

Hydrogen evolution kinetics during water decomposition for electrodes: non-activated (1), activated in 1 М КОН (2) and 0.5 М Na2SO4 (3), upon illumination with LED-light with the wavelength 400 nm.
The process of the TiO2-nanotubes’ electrochemical doping accelerated the hydrogen evolution as compared with the initial sample. For example, in the water electrolysis with non-activated TiO2 the hydrogen evolution rate was 0.22 μmol min–1 Н2, while upon the CV-activation the Н2 evolution rate increased by a factor of 5.4–6.8 and came to 1.2 and 1.5 μmol min–1 for the electrodes processed in 1 М КОН and 0.5 М Na2SO4 respectively.
The electrochemical impedance spectroscopy is widely used in the studying of the electrical properties of semiconductors, as well as the mechanism and kinetics of processes occurring at the electrode/electrolyte interface. In Fig. 5 we show experimental electrochemical-impedance-spectroscopy data as Nyquist diagrams for the electrodes prior to and after the electrochemical doping. The latter show frequency dependence of the impedance in the coordinates of the impedance imaginary and real components. We analyzed the obtained impedance spectra by using the equivalent circuit presented schematically in the Insert to Fig. 5.

Nyquist diagram obtained for electrodes: non-activated (1), activated in 1 М КОН (2) and 0.5 М Na2SO4 (3), upon illumination with light with the wavelength λ: 400 nm (а), 450 nm (b).
The obtained impedance spectra were analyzed using this equivalent circuit model. The semicircles at moderate frequencies in the Nyquist diagrams contain information on the charge transfer processes at the electrode/electrolyte interface. At that, the semicircle diameters projected onto the impedance real-part axis correspond to the charge-transfer resistance [22]. The cyclic-voltammetry activation leads to a significant lowering of the charge transfer resistance, which manifested itself in a decrease of the semicircle diameter.
In Table 2 we give values of the equivalent circuit parameters obtained upon the best convergency with the model.
Upon the irradiation with the light with wavelength 400 nm the full charge transfer resistance Rfull for the electrochemically activated electrodes came to 185.7–275.9 Ω, while for the initial sample this quantity exceeded 13 kΩ. At λ = 450 nm, Rfull is 189.5, 5 and 12.6 kΩ for the non-activated and reduced in 1 М КОН and 0.5 М Na2SO4 electrodes, respectively. The resistance lowering upon the electrochemical doping is reached because of the increase in the oxide-layer volume capacitance. This leads to larger charge density at the oxide/solution interface, hence, to the corresponding increase in the electrical double layer capacitance and decrease in the charge transfer resistance [16, 19, 23]. In addition, for the electrochemically reduced samples, an increase of the surface states’ density is possible. The surface states is an important path for the electron transfer, which favored the charge separation and increase in the samples’ photoactivity [19].
CONCLUSIONS
Nanostructured anodic TiO2 films were activated by the cyclic voltammetry method. The electrochemical reductive doping increased the photoelectrocatalytical activity of the electrodes upon UV-A-irradiation (λ = 400 nm) and visible light (λ = 450 nm) illumination. At the electrodes activated by the cyclic-voltammetry method, the photocurrent density increased by a factor of 4 to 7 upon the visible light illumination; 5–14, upon the UV-A-irradiation (depending on the applied potential) as compared with non-treated sample. The electrochemical activation is shown to increase drastically the hydrogen evolution rate when the UV-A-irradiation has been used.
It was shown by the electrochemical impedance spectroscopy method that TiO2-nanotubes’ electrochemical hydration resulted in a drastic decrease of the charge transfer resistance at the electrode/electrolyte interface and increase in the oxide layer electrical conductivity. This favors the charge-carrier separation, decrease in the electron-hole pair recombination rate and, therefore, increase in the anode photoactivity. It was shown that the cyclic voltammetry could be used as an effective method for the improvement of photoelectrochemical characteristics of the water splitting process.
REFERENCES
Chen, X., Liu, L., Yu, P.Y., and Mao, S.S., Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals, Science, 2011, vol. 331, p. 746 .
Ying, D., Cao, R., Li, C., Tang, T., Li, K., Wang, H., Wang, Y., and Jia, J., Study of the photocurrent in a photocatalytic fuel cell for wastewater treatment and the effects of TiO2 surface morphology to the apportionment of the photocurrent., Electrochim. Acta, 2016, vol. 192, p. 319.
Li, H., Chen, Z., Tsang, C. K., Li, Z., Ran, X., Lee, C., Nie, B., Zheng, L., Hung, T.F., Lu, J., Pan, B., and Li, Y., Electrochemical doping of anatase TiO2 in organic electrolytes for high-performance supercapacitors and photocatalysts, J. Mater. Chem., 2014, vol. 2, p. 229.
Palmas, S., Polcaro, A.M., Ruiz, J.R., Pozzo, A.D., Mascia, M., and Vacca, A., TiO2 photoanodes for electrically enhanced water splitting, Int. J. Hydrogen Energy, 2010, vol. 35, p. 6561.
Sahoo, S. S., Mansingh, S., Babu, P., and Parida, K., Black titania an emerging photocatalyst: review highlighting the synthesis techniques and photocatalytic activity for hydrogen generation, Nanoscale Advances, 2021, vol. 3, p. 5487.
Samioloa, L., Valigi, M., Gazzoli, D., and Amadelli, R., Photo-electro catalytic oxidation of aromatic alcohols on visible light-absorbing nitrogen-doped TiO2, Electrochim. Acta, 2010, vol. 55, no. 26. p. 7788.
Xing, M.-Y., Li, W.-K., Wu, Y.-M., Zhang, J.-L., and Gong, X.-Q., Formation of New Structures and Their Synergistic Effects in Boron and Nitrogen Codoped TiO2 for Enhancement of Photocatalytic Performance, J. Phys. Chem. C, 2011, vol. 115, no. 16, p. 7858.
Naldoni, A., Altomare, M., Zoppellaro, G., Liu, N., Kment, Š., Zbořil, R., and Schmuki, P., Photocatalysis with Reduced TiO2: From Black TiO2 to Cocatalyst-Free Hydrogen Production, ACS Catalysis, 2019, vol. 9, p. 345.
Wang, Z., Wang, Z., Yang, C., Yang, C., Lin, T., Lin, T., Yin, H., Chen, P., Wan, D., Xu, F., Huang, F., Huang, F., Lin, J., Xie, X., and Jiang, M., Visible-light photocatalytic, solar thermal and photoelectrochemical properties of aluminium-reduced black titania, Energy Environmental Sci., 2013, vol. 6, p. 3007.
Shah, M., Zhu, Y., Fan, X., Zhao, J., Li, Y., Asim, S., and Wang, C., Facile Synthesis of Defective TiO2–x Nanocrystals with High Surface Area and Tailoring Bandgap for Visible-light Photocatalysis, Sci. Reports, 2015, vol. 5, p. 15804.
Li, G., Lian, Z., Li, X., Xu, Y., Wang, W., Zhang, D., Tian, F., and Li, H., Ionothermal synthesis of black Ti3+-doped single-crystal TiO2 as an active photocatalyst for pollutant degradation and H2 generation, J. Mater. Chem. A, 2015, vol. 3, p. 3748.
Panomsuwan, G., Watthanaphanit, A., Ishizaki, T., and Saito, N., Water-plasma-assisted synthesis of black titania spheres with efficient visible-light photocatalytic activity, Phys. Chem. Chem. Phys., 2015, vol. 17, p. 13794.
Nakajima, T., Nakamura, T., Shinoda, K., and Tsuchiya, T., Rapid formation of black titania photoanodes: pulsed laser-induced oxygen release and enhanced solar water splitting efficiency, J. Mater. Chem. A, 2014, vol. 2, no. 19, p. 6762.
Yang, Y., Liao, J., Li, Y., Cao, X., Li, N., Wang, C., and Lin, S., Electrochemically self-doped hierarchical TiO2 nanotube arrays for enhanced visible-light photoelectrochemical performance: an experimental and computational study, RSC Advances, 2016, vol. 6, no. 52, p. 46871.
Wu, H., Li, D., Zhu, X., Yang, C., Liu, D., Chen, X., Song, Y., and Lu, L., High-performance and renewable supercapacitors based on TiO2 nanotube array electrodes treated by an electrochemical doping approach, Electrochim. Acta, 2014, vol. 116, p. 129.
Fabregat-Santiago, F., Barea, E. M., Bisquert, J., Mor, G.K., Shankar, K., and Grimes, C.A., High carrier density and capacitance in TiO2 nanotube arrays induced by electrochemical doping, J. Amer. Chem. Soc., 2008, vol. 130 34, p. 11312.
Berger, T., Lana-Villarreal, T., Monllor-Satoca, D., and Gómez, R., Charge transfer reductive doping of nanostructured TiO2 thin films as a way to improve their photoelectrocatalytic performance, Electrochem. Commun., 2006, vol. 8, no. 11, p. 1713.
Jankulovska - Petkovska, M., Berger, T., Wong, S., Gómez, R., and Lana-Villarreal, T., Trap States in TiO2 Films Made of Nanowires, Nanotubes or Nanoparticles: An Electrochemical Study, Chemphyschem: Europ. J. Chem. Phys. Phys. Chem., 2012, vol. 13, p. 3008.
Zhu, H., Zhao, M., Zhou, J., Li, W., Wang, H., Xu, Z., Lu, L., Pei, L., Shi, Z., Yan, S., Li, Z., and Zou, Z., Surface states as electron transfer pathway enhanced charge separation in TiO2 nanotube water splitting photoanodes, Appl. Catal. B: Environmental, 2018, vol. 234, p. 100.
Varghese, O.K. and Grimes, C.A., Appropriate strategies for determining the photoconversion efficiency of water photoelectrolysis cells: A review with examples using titania nanotube array photoanodes, Solar Energy Mater. Solar Cells, 2008, vol. 92, no. 4. p. 374.
Yu, D., Zhang, Y., Wang, F., and Dai, J., Preparation of ZnO/two-layer self-doped black TiO2 nanotube arrays and their enhanced photochemical properties, RSC Advances, 2021, vol. 11, no. 4, p. 2307.
Bredar, A., Chown, A., Burton, A., and Farnum, B., Electrochemical Impedance Spectroscopy of Metal Oxide Electrodes for Energy Applications, ACS Appl. Energy Mater., 2020, vol. 3, p. 66.
Leshuk, T., Parviz, R., Everett, P., Krishnakumar, H., Varin, R. A., and Gu, F. X., Photocatalytic activity of hydrogenated TiO2, ACS Appl. Mater. Interfaces, 2013, vol. 56, p. 1892.
Funding
This work is carried out under the State Contract of the Federal Research Center, Krasnoyarsk Scientific Center, Siberian Branch, RAS (project no. 0287-2021-0023), with the using of equipment of the Krasnoyarsk Research Equipment Sharing Center of the Federal Research Center, Krasnoyarsk Scientific Center, Siberian Branch, RAS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by Yu. Pleskov
Delivered at the 20th All-Russian Meeting “Electrochemistry of Organic Compounds” (EKhOS-2022), Novocherkassk, October 18–22, 2022.
Rights and permissions
About this article

Cite this article
Zos’ko, N.A., Kenova, T.A., Taran, O.P. et al. Electrochemical Reductive Doping of TiO2-Nanotubes to Increase the Efficiency of Photoelectrochemical Water Splitting. Russ J Electrochem 59, 767–773 (2023). https://doi.org/10.1134/S1023193523100142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1023193523100142