
Abstract
The electrochemical characteristics of carbonaceous materials differing in their specific surface area and porosity are studied by the method of cyclic voltammetry in 0.25 М LiClO4/DMSO electrolyte, both in inert and oxygen atmosphere. The value of the electrochemically active surface area that was estimated from cyclic voltammograms as the polarization capacitance was shown to increase with increase in the BET specific surface area. The efficiency in the oxygen reaction, measured in the oxygen atmosphere and expressed as the charge consumed for the formation of Li2O2 (QC) in the oxygen reduction reaction (the cathodic segment), and the process reversibility, expressed as the ratio of the charge consumed for oxygen evolution (QA) (the anodic segment) to QC, are mainly determined by the electrochemically active surface area and the porosity of the carbonaceous material.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The development of positive electrodes for the Li–O2-current sources involved different carbonaceous materials: carbon blacks, carbon nanotubes, carbon fibers, graphenes, etc., characterized by different specific surface areas and porous structures [1, 2]. To provide maximal area of the electrochemically-active cathode surface, the usage of highly dispersive materials is preferable. The carbonaceous materials approach closely to the optimum, thanks to their good availability, low price, and easy formation of preset structures.
When oxygen is reduced at positive electrode of a lithium–oxygen current source (here, functioning as a cathode), solid Li2O2 accumulates and blocks the electrode active surface and lowers the pores’ free volume and surface area. Ultimately, this results in the pore clogging and makes the oxygen transport more complicated. It also complicates the lithium ion and electron transfer, thus lowering the current source entire capacity considerably. To reach the Li–O2-current source characteristics approaching the theoretical ones, of necessity is to keep optimal transport regime. This is precisely why the understanding of the used carbonaceous materials effect on the positive electrode characteristics is one of central problems.
The interdependence of the discharge capacity and the positive electrode structure was studied in aprotic solvents (DME, tetraglyme, DMSO, etc.) [2–6]. The interrelation between the specific discharge capacity and a carbonaceous material surface area was elucidated in work [4]. As opposed thereto, it was shown [5] that macroporous nanofoams (with the pore diameter 50–200 nm), despite their lower surface area, demonstrated twice as much capacity as compared with the mesoporous ones (the pore diameter 5–50 nm). The discharge capacity was shown [2] to have no considerable dependence on the carbonaceous material’s specific surface area or the pore volume; it is solely determined by the pore size. In work [8], a composite cathode consisting of carbon black and carbon nanotubes was studied. The system was shown to have larger capacity than cathodes based on the carbon black only, despite the fact that the mixture had lower specific surface area. The effect was explained [8] by the intensifying of oxygen transport in thick-pore materials. We see that currently there is no consensus on the effect of the carbonaceous material surface area and porosity on the Li–O2-current sources characteristics.
In addition to the pore size and volume, considerable impact on the current source characteristics is likely to be exerted also by the pore distribution in size over the electrode active layer thickness. The oxygen reduction reaction in the Li+-containing electrolyte can be generally written as:
During the discharge, the oxygen concentration in the porous cathode bulk decreased in the direction from the gas input to the cathode/electrolyte contact. The oxygen higher concentration provided the formation of a Li2O2 larger amount. On this reason, the amount of the solid lithium peroxide formed in the cathodic layer increased at the side of the O2 input and gradually decreased toward the electrode/electrolyte contact. This result found experimental confirmation [7] and was also confirmed by digital simulation [8‒10]. With this type of the solid product accumulation, the main part of the pores at the side of the O2 delivery, evidently, is blocked and cannot help but take a toll on the decrease of the battery entire discharge capacity. At that, thin cathodes cannot solve the problem because they cannot provide large electrochemically active surface area, hence, large amount of Li2O2. The above-described scenario allows deeming advisable formation of thick cathodic layers in which the pore size decreased from the side of the O2 delivery toward the electrolyte boundary. This is possible, e.g., in the layer-by-layer manufacturing, with the using of carbonaceous materials of different porosity, as well as their mixtures. This approach is likely to provide more uniform and stable oxygen distribution, which has been favorable for the current source discharge capacity.
The acceleration of the current-producing reactions and decreasing of their overvoltage is of great importance for successful implementation of the entire Li–O2-battery charging–discharging cycle. The choice of catalysts for the Li–O2-current sources is a more complicated task as compared with, e.g., Н2–О2-fuel cells, because the electrode materials for the positive electrode must provide electrocatalysis of both oxygen reduction in the presence of Li+ and oxygen evolution reactions. Different researchers suggested both bifunctional systems [11] and composed electrodes comprising two independent electrodes [12, 13]. Also, metal-modified carbonaceous materials are under consideration. It is to be noted that the stability of the positive electrode materials is of great significance for the Li–O2-current sources, the more so, in their charging and discharging (cycling).
The bifunctional catalysts for the positive electrodes in nonaqueous solutions can be carbonaceous materials modified with different metals, e.g., Pt [14], Au [15], Pd [16], Co [17], etc. There exist original ideas for the structural implementation of metal–carbonaceous support pairs. For example, a system was developed [18] in which Pt, Pd, Ru, and Au nanoparticles were placed edgewise to carbon nanotubes. Such cathodic materials demonstrated significantly lower overvoltage during the lithium–oxygen battery charging as compared with their analogs (the metals deposited onto the carbon nanotube’ surface). It was shown in the work [19] devoted to disperse cathodic systems of the type of M1/С, М1М2/С (where М1 and М2 is the metal; С is the carbonaceous material), as well as oxides and different combined catalysts, that, unlike uncoated carbonaceous materials, the catalytic effects of such materials manifest themselves mainly during the charging and is weakly pronounced or even absent in the oxygen reduction in the presence of Li+.
In spite of a great number of works devoted to the above-described topic, the mechanisms of the current-producing charging–discharging processes in the Li–O2-current sources are still understudied and the interpreting of obtained data appears being not unambiguous. The mere usage of polarization curves obviously is an important, yet, insufficient tool for the full understanding of the reaction paths and the occurring processes’ character. In particular, in work [20] differential electrochemical mass-spectroscopy was applied to the studying of charging processes. The noble-metal-modification (e.g., with Pt, Ru, etc.) was shown not to provide the expected process acceleration. The modifications promoted СО2 evolution as a result of the carbonaceous materials’ degradation, rather than the oxygen evolution reaction intensifying; thus, the current sources reversibility decreased. This unambiguously points to the necessity of the studying more extensively the catalytic systems, for objective appraisal of their influencing the processes occurring in the positive electrode of lithium–oxygen batteries.
The advantages of carbonaceous materials in the Li–O2-current sources are as follows: large specific surface area, porous structure, activity, and apparent economic value. Here, it is understood that the materials are surrounded by aggressive oxidative medium and are prone to the influence of the Li2O2 decomposition products, which can destroy the materials [21]. The carbonaceous materials are shown [22] to be stable at potentials below 3.5 V; they degraded during charging at still higher potentials (~4.0 V).
In this work, we aimed at the studying of the carbonaceous materials’ effectiveness as active layer materials for positive electrodes of different morphology in the 0.25 М LiClO4/DMSO-electrolyte, as well as elucidating of interrelation between their structural characteristics and their activity in the oxygen reduction and evolution reactions.
EXPERIMENTAL
The studies were performed in electrolyte based on DMSO-solvent and lithium salt LiClO4 at a concentration of 0.25 М. Prior to the electrolyte preparation, the LiClO4 anhydrous salt was subjected to additional drying in a vacuum desiccator at 80°С. The measurements were carried out at 20°С in hermetic three-electrode glass cell. The electrode was a stationary disc of glassy carbon, with the working surface diameter of 4.0 mm (the surface area 0.126 сm2). Prior to the measurements, the cell was dried carefully, blown-through with argon, and filled with the electrolyte in a dry glove-box; afterwards, the measurements were carried out in air. The water content in the electrolyte was determined after Fischer; it was below 100 ppm. The reference electrode was a silver wire immersed into 0.01 М AgNO3 solution. To measure the working electrode potential relative to the Li/Li+ couple, the reference Ag/Ag+-electrode was calibrated in DMSO vs. lithium electrode. The calibration was performed by using the silver wire immersed into 0.01 М AgNO3 solution and lithium immersed into 0.25 М LiClO4 solution in DMSO. The electrodes were interconnected by electrolytic bridge filled with a lithium-salt solution. The measurements were carried out in a dry glove-box. Under the chosen conditions, the silver electrode potential is: ЕAg/Ag+ = 3.58 V (Li/Li+). This value was used in the recalculating of the working electrode potential measured vs. Ag/AgNO3 into the Li/Li+-electrode scale of potentials.
In this work, we used commercial carbonaceous materials: carbon blacks (Super P, ХС72, EC600JD), carbon nanotubes (Taunit-М, Tambov), and carbon nanoflakes. The carbon nanoflakes’ structure and synthesis were described at length elsewhere [23]. The above-listed carbonaceous materials differ both in their pore size and the specific surface area; the latter was estimated for the novel materials (carbon nanotubes and carbon nanoflakes) by the BET-method, while literature information [24] was used for the commercial samples.
To deposit the studied disperse materials onto the electrode, we prepared “ink”, that is, a suspension of a carbonaceous material powder in isopropanol (in all cases, 2 mg in 500 μL of isopropanol). The uniformity of the deposition onto the electrode is promoted by the introducing of Nafion 5%-solution in a mixture of low-molecular aliphatic alcohols (Aldrich, 0.03 mL), diluted by a factor of ten. The suspension was sonicated in baths for 30–40 min. It was deposited onto the electrode by a micropipette (4 μg of a carbonaceous material sample in 1 μL). The electrodes were dried in air. The method provided the electrode loading by the carbonaceous materials of ~100 μg/сm2.
The measurements were carried out at a temperature of 20°С in the electrolyte 0.25 М LiClO4/DMSO. Cyclic voltammograms were recorded in the argon-deaerated (for 30 min) solution at a potential scanning rate of 50 mV/s. The electrochemically active surface area (SEA, mC/cm2) was estimated by the charge that characterized the polarization capacity; the latter was determined by the integrating of the cyclic-voltammogram anodic segment. Then the solution was saturated with oxygen for 30 min, and current-voltage curves were recorded over the voltage range of 2.0–4.0 V (Li/Li+) at a potential scanning rate of 5 mV/s. To calculate the charge consumed in the reduction or oxidation, the segments under the curves (the direct and reverse runs) were integrated separately, then the Q values were summated and the sought for values ΣQC, ΣQА have been obtained. The oxygen reaction reversibility was obtained from the ΣQА/ΣQC ratio. The calculations’ scheme was described elsewhere [24].
RESULTS AND DISCUSSION
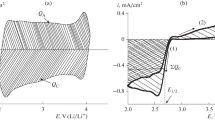
The electrochemically active surface area (SEA) of the carbonaceous materials was estimated from the cyclic voltammograms taken for the carbon blacks Super P, ХС72, EC600JD, as well as for the carbon nanotubes and carbon nanoflakes. In Fig. 1 we give a typical cyclic voltammogram; in Fig. 2, values of the electrochemically active surface area and the BET data for all carbonaceous materials studied.

Cyclic voltammograms taken at different carbonaceous materials; 50 mV/s, 20°С, 0.25 М LiClO4/DMSO, Ar atmosphere. The carbonaceous material load at the electrode: 100 μg/сm2.

Dependence of the charge determined from cyclic voltammograms on the BET-surface area. 20°С, 0.25 М LiClO4/DMSO, Ar atmosphere. The carbonaceous material (carbon nanotubes and carbon nanoflakes) load at the electrode: 100 μg/сm2.
We correlated the specific surface area found for the studied samples after the BET method and the charge obtained by the cyclic voltammogram integrating. With the increasing of the BET surface area by a factor of ~20 (from 60 up to 1270 m2 g–1) when passing from the Super P carbon black to EC600JD, we observed the increase of SEA by a factor of ~10, which evidenced the underuse of the carbonaceous material specific surface area. Not all of the carbonaceous material surface area is electrochemically active because of nonuniformity of the inner surface lyophilic/lyophobic properties in the porous samples. Not all spots on the surface are uniformly wetted by the electrolyte, and this resulted in inadequate SEA growth with the increasing of SBET. In addition, the thickness of the deposited layer (100 μg/сm2) differs because of the difference in the carbonaceous materials’ density, and this contributes to the electrochemically active surface area measured at a potential scanning rate of 50 mV/s.
In Fig. 3 we present polarization curves of the oxygen reaction obtained at the carbonaceous materials in oxygen atmosphere. In Table 1 we summarized the results of the studies. The Super P carbon black consists of chains of spheroids sized ~40 nm, whose bulk layer forms open structures with macropores sized up to 500 nm. The Super P specific surface area is the lowest among the carbonaceous materials studied; it came to 60 m2 g–1 [25]. For the carbon nanoflakes with the specific surface area after BET above 1200 m2 g–1, the volume fraction occupied by the micropores is larger, which already has been noted in work [23]; therefore, in what follows, a sample of carbon nanoflakes with the specific surface area of 1180 m2 g–1 was used as a ground sample.

Oxygen reduction at different carbonaceous materials. 5 mV/s, 20°С, 0.25 М LiClO4/DMSO, О2 atmosphere. The carbonaceous material load at the electrode: 100 μg/сm2.
In Fig. 4, the reaction reversibility (ΣQA/ΣQC) is presented as a function of the carbonaceous material specific surface area. With the increasing of the carbonaceous material specific surface area (measured by the BET method) by a factor of 6, we observed the reversibility increase by a factor of 2, on the average. The increase in the reversibility with the increasing of the specific surface area is likely to be entirely caused by the extensive growth of the electrochemically active surface area; it is not connected with any catalytic effect. The Li2O2 is formed at a carbonaceous material with developed surface as disjoint islets which are oxidized during the anodic run at a lower overvoltage, as compared with carbonaceous materials with lower specific surface area, at which the Li2O2 formation can lead to deposition of several lithium peroxide monolayers, hence, the surface blocking. The oxidation of such a layer during the anodic run is hindered or even became quite impossible.

The reversibility (ΣQA/ΣQC) of oxygen reaction at different carbonaceous materials. 20°С, 0.25 М LiClO4/ DMSO, О2 atmosphere. The carbonaceous material load at the electrode: 100 μg/сm2.
This idea is confirmed by the ΣQA/ΣQC dependence on the carbonaceous-material load at the electrodes (the load ranged from 25 to 100 μg, Fig. 5). When comparing a mesoporous sample with moderate surface area (carbon nanotubes) and micro- and mesoporous materials with larger surface area (carbon nanoflakes and EC600JD), we observed that with the increasing of the loading the character of the ΣQA/ΣQC growthg is different. The increasing of the carbon nanotubes’ loading by a factor of 4 resulted in but negligible increase of the reversibility. Similar effect was observed for the EC600JD whose surface area, being composed by the micropore bulk, is not fully electrochemically active. In the case of carbon nanoflakes, the increase of the loading by a factor of 4 resulted in the increase of the reversibility by 60–70%. According to the BET data and work [23], in the carbon nanoflakes over 50% of their pore volume relates to the pore size of 10–35 nm. This fact has fueled the suggestions that the inner volume of the pores participates more fully in the electrochemical reaction. On the other hand, the value of QC for the studied carbonaceous materials makes little difference as compared with the factor ΣQA/ΣQC, which increased appreciably just with the increasing of SEA. When the surface SEA is incompletely blocked by Li2O2, hence, some spots for the electron transfer are at hand, some favorable conditions are likely to be created for the lowering of the lithium-peroxide oxidation overvoltage.

Dependence of the reaction reversibility ΣQA/ΣQC on the carbonaceous material load at the electrode. 20°С, 0.25 М LiClO4/DMSO, О2 atmosphere.
It is the reaching of large overall current that justified the usage of the cathodes with increased carbonaceous-material loading. However, because of side-products (Li2O2, LiO2, Li2CO3) formation, the using of thick cathodic layers with microporous structure is fraught with the danger of the micropores’ complete abandonment because of their slugging with Li2O2. In Fig. 6 we counterposed the 1st and 5th cycles in the polarization curves taken with the carbon nanoflakes and Super P. Upon the multiple cycling over the studied potential range, the Super P carbon black (that is. a material with lower SEA value) demonstrated the reversibility downfall below 0.2, whereas for the carbon nanoflakes (a mesoporous material with larger SEA) the ΣQA/ΣQC value retained this factor over 0.5. Evidently, the increased gas permeability of the layer in the case of the using of carbonaceous materials with the high specific surface area and SEA favors the positive electrode operational stability. Thus, the mesoporous structure is the factor that provided effective transportation of the reagents of electrochemical reactions to active centers at the positive-electrode material (carbon nanotubes and carbon nanoflakes) surface. Here, the large surface area favors the lithium peroxide accumulation as islets and Li2O2-free spots. Thereby, electrons are easily transported to the lithium-peroxide-free zone; yet, it can be oxidized at neighbor spots. As a result, the charging overvoltage is lowered.

Oxygen reduction at Super P (1, 1 ′) and carbon nanoflakes (2, 2 ′). 1st cycle (1, 2); 5th cycle (1 ', 2 '). 5 mV/s, 20°С, 0.25 М LiClO4/DMSO, О2 atmosphere.
CONCLUSIONS
The oxygen-reaction activity and reversibility are studied in aprotic electrolyte LiClO4/DMSO at carbonaceous materials which differ in their specific surface area and porosity. Among the studied materials, most perspective for further studies must be carbon nanotubes and carbon nanoflakes, that is, the carbonaceous materials whose structure provides more efficient using of the specific surface area.
REFERENCES
Kraytsberg, A. and Ein-Eli, Y., Review on Li–air batteries—Opportunities, limitations and perspective, J. Power Sources, 2011, vol. 196, p. 886.
Ding, N., Chien, S.W., Hor, T.S.A., Lum, R., Zong, Y., and Liu, Z., Influence of carbon pore size on the discharge capacity of Li–O2 batteries, J. Mater. Chem. A., 2014, vol. 2, p. 12433.
Kim, M., Yoo, E., Ahn, W.-S., and Shim, S.E., Controlling porosity of porous carbon cathode for lithium oxygen batteries: Influence of micro and meso porosity, J. Power Sources, 2018, vol. 389, p. 20.
Meini, S., Piana, M., Beyer, H., Schwaemmlein, J., and Gasteiger, H.A., Effect of carbon surface area on first discharge capacity of Li–O2 cathodes and cycle-life behavior in ether-based electrolytes, J. Electrochem. Soc., 2012, vol. 159, p. A2135.
Chervin, C.N., Wattendorf, M.J., Long, J.W., Kucko, N.W., and Rolison, D.R., Carbon nanofoam-based cathodes for Li–O2 batteries: correlation of pore solid architecture and electrochemical performance, J. Electrochem. Soc., 2013, vol. 160, p. A1510.
Tan, P., Shyy, W., Wei, Z.H., An, L., and Zhao, T.S., A carbon powder-nanotube composite cathode for non-aqueous lithium-air batteries, Electrochim. Acta, 2014, vol. 147, p. 1.
Zhang, G.Q., Zheng, J.P., Liang, R., Zhang, C., Wang, B., Hendrickson, M., and Plichta, E.J., Lithium–air batteries using SWNT/CNF buckypapers as air electrodes, J. Electrochem. Soc., 2010, vol. 157, p. A953.
Sahapatsombut, U., Cheng, H., and Scott, K., Modelling the micro–macro homogeneous cycling behaviour of a lithium–air battery, J. Power Sources, 2013, vol. 227, p. 243.
Yoo, K., Banerjee, S., and Dutta, P., Modeling of volume change phenomena in a Li–air battery, J. Power Sources, 2014, vol. 258, p. 340.
Jung, C.Y., Zhao, T.S., and An, L., Modeling of lithium–oxygen batteries with the discharge product treated as a discontinuous deposit layer, J. Power Sources, 2015, vol. 273, p.440.
Wang, L., Zhao, X., Lu, Y., Xu, M., Zhang, D., Ruoff, R.S., Stevenson, K.J., and Goodenough, J.B., CoMn2O4 spinel nanoparticles grown on graphene as bifunctional catalyst for lithium-air batteries, J. Electrochem. Soc., 2011, vol. 158, p. A1379.
Li, L. and Manthiram, A., Decoupled bifunctional air electrodes for highperformance hybrid lithium-air batteries, Nano Energy, 2014, vol. 9, p. 94.
Li, L., Liu, C., He, G., Fan, D., and Manthiram, A., Hierarchical pore-in-pore and wire-inwire catalysts for rechargeable Zn– and Li–air batteries with ultra-long cycle life and high cell efficiency, Energy Environ Sci., 2015, vol. 8, p. 3274.
Lim, H.D., Song, H., Gwon, H., Park, K.-Y., Kim, J., Bae, Y., Kim, H., Jung, S.-K., Kim, T., Kim, Y.H., Lepró, X., Ovalle-Robles, R., Baughmand, R.H., and Kang, K., A new catalyst-embedded hierarchical air electrode for high-performance Li–O2 batteries, Energy Environ. Sci., 2013, vol. 6, p. 3570.
Marinaro, M., Riek, U., Moorthy, S.K.E., Bernhard, J., Kaiser, U., Wohlfahrt-Mehrens, M., and Jörissen, L., Au-coated carbon cathodes for improved oxygen reduction and evolution kinetics in aprotic Li–O2 batteries, Electrochem. Commun., 2013, vol.37, p. 53.
Zhu, D., Zhang, L., Song, M., Wang, X., and Chen, Y., An in situ formed Pd nanolayer as a bifunctional catalyst for Li–air batteries in ambient or simulated air, Electrochem. Commun., 2013, vol. 49, p. 9573.
Zhang, Z., Su, L., Yang, M., Hu, M., Bao, J., Wei, J., and Zhou, Z., A composite of Co nanoparticles highly dispersed on N-rich carbon substrates: an efficient electrocatalyst for Li–O2 battery cathodes, Chem. Commun., 2014, vol. 50, p. 776.
Huang, X., Yu, H., Tan, H., Zhu, J., Zhang, W., Wang, C., Zhang, J., Wang, Y., Lv, Y., Zeng, Z., Liu, D., Ding, J., Zhang, Q., Srinivasan, M., Ajayan, P.M., Hoon, H., and Yan, Q., Carbon nanotube encapsulated noble metal nanoparticle hybrid as a cathode material for Li–oxygen batteries, Adv. Funct. Mater., 2014, vol. 24, p. 6516.
Tan, P., Jiang, H.R., Zhu, X.B., An, L., Jung, C.Y., Wu, M.C., Shi, L., Shyy, W., and Zhao, T.S., Advances and challenges in lithium–air batteries, Applied Energy, 2017, vol. 204, p. 780.
Ma, S., Wu, Y., Wang, J., Zhang, Y., Zhang, Y., Yan, X., Wei, Y., Liu, P., Wang, J., Jiang, K., Fan, S., Xu, Y., and Peng, Z., Reversibility of noble metal-catalyzed aprotic Li–O2 batteries, Nano Lett., 2015, vol. 15, p. 8084.
McCloskey, B.D., Speidel, A., Scheffler, R., Miller, D.C., Viswanathan, V., Hummelshoj, J.S., Nørskov, J.K., and Luntz, A.C., Twin problems of interfacial carbonate formation in nonaqueous Li–O2 batteries, J. Phys. Chem. Lett., 2012, vol. 3, p. 997.
Itkis, D.M., Semenenko, D.A., Kataev, E.Yu., Belova, A.I., Neudachina, V.S., Sirotina, A.P., Hävecker, M., Teschner, D., Knop-Gericke, A., Dudin, P., Barinov, A., Goodilin, E.A., Shao-Horn, Y., and Yashina, L.V., Reactivity of carbon in lithium–oxygen battery positive electrodes, Nano Lett., 2013, vol. 13, p. 4697.
Arkhipova, E.A., Ivanov, A.S., Maslakov, K.I., Egorov, A.V., Savilov, S.V., and Lunin, V.V., Mesoporous graphene nanoflakes for high performance supercapacitors with ionic liquid electrolyte, Microporous Mesoporous Materials, 2020, vol. 294, p. 109851.
Bogdanovskaya, V.A., Panchenko, N.V., Radina, M.V., Andreev, V.N., Korchagin, O.V., Tripachev, O.V., and Novikov, V.T., Oxygen reaction at carbonaceous materials with different structure in electrolytes based on lithium perchlorate and aprotic solvents, Russ. J. Electrochem., 2019, vol. 55, p. 878.
Olivares-Marín, M., Aklalouch M., and Tonti, D., Combined influence of meso- and macroporosity of soft-hard templated carbon electrodes on the performance of Li–O2 cells with different configurations, Nanomaterials, 2019, vol. 9, p. 810.
Funding
This work was financially supported by the Ministry of Sciences and Higher Education of the Russian Federation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by Yu. Pleskov
Rights and permissions
About this article

Cite this article
Kuzov, A.V., Bogdanovskaya, V.A., Emets, V.V. et al. The Effect of Carbonaceous Material Morphology on Oxygen Reduction Reaction in Nonaqueous Electrolyte Containing Lithium Ions. Russ J Electrochem 58, 296–302 (2022). https://doi.org/10.1134/S1023193522040073
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1023193522040073