
Abstract
The electroreduction (ER) of benzo[a]phenazine-7,12-dioxide (1) and 2-ethoxycarbonyl-3-methyl-quinoxaline-1,4-dioxide (2) in DMF on a glassy carbon electrode is studied by the methods of cyclic voltammetry, chronoamperometry, and electrolysis at controlled potential. In aprotic medium, these compounds are reduced to form relatively stable complexes as observed in both cyclic voltammetry curves and UV spectra. The deoxygenation of the derivatives of phenazine and quinoxaline N,N′-dioxides proceeds as a result of decomposition of the radical formed either at the ER of complexes of these compounds with СН3СООН or as a result of protonation of radical anions. For compound 2, the competition between the reactions of decomposition and ER of this radical is observed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The derivatives of N,N′-dioxides of phenazine and quinoxaline are often considered [1–3] as the potential medicinal drugs for curing “hard” tumors with the large hypoxic regions where almost no oxygen can penetrate. Under hypoxia conditions, the cells become resistant to radiation and chemotherapy. The conventional chemotherapeutic drugs act on the rapidly growing carcinogenic cells thus preventing their fission and, hence, are inefficient for cells under hypoxia conditions because the latter divide very slowly. The new approaches to antitumoral therapy are being developed on the basis of the so-called prodrugs [4, 5], which are reduced by enzymes in vivo under hypoxia conditions to form drugs capable of affecting carcinogenic cells. The derivatives of heteroaromatic N-oxides pertain exactly to these bioreducible compounds. Tirapazamine, i.e., 1,3-amino-1,2,4-benzotriazine-1,4-N-dioxide [6, 7], is one of the first drugs with high cytotoxicity toward cells under hypoxia conditions to be introduced into the clinical practice. By using various biological research techniques it was shown [8, 9] that the one-electron reduction of tirapazamine by enzymes produces the highly reactive radical •OH that destroys the DNA of tumor cells. It is generally assumed that the process of prodrug deoxygenation is the most efficient exactly under the conditions of low oxygen levels because oxygen prevents the reduction processes in tissues. The questions on the mechanism of redox reactions of prodrugs and interaction of produced radicals with the DNA are still actively discussed in the literature [9].
In recent years, the phenazine derivatives were considered as the most promising compounds for the synthesis of prodrugs with the high antitumoral activity [10, 11]. The formation of the •OH radical in the bioreduction of derivatives of phenazine dioxides, as in tirapazamine bioreduction, is associated with the biological activity of dioxides. However, considering the behavior of derivatives of phenazine dioxides containing various substituents made it possible to conclude that this radical is formed not in all the cases [4]. To systematize the information on the potentially active compounds, attempts were undertaken to find the relationship between the antitumoral activity and the physicochemical properties of compounds, particularly, with their reduction potential [12, 13]. Far from all structures of substituted phenazine N,N'-dioxides obey such linear correlation; probably, their reduction mechanism (or their effect on the cells) are substantially different for structures with different substituents. Insofar as the formation of radical species is associated with the redox properties of heteroaromatic N,N'-dioxides, then studying the electron-transfer stages and near-electrode chemical reactions by electrochemical techniques may be useful for determining the criteria of assessing the prospects of organic compounds synthesized.
In this study, to obtain information on the mechanism of reduction of aromatic dioxides, we used cyclic voltammetry (CV), chronoamperometry (CA), and electrolysis at controlled potential (ECP). As the model compounds for studying the electroreduction (ER) of derivatives of N,N'-dioxides of phenazine and quinoxaline, we used benzo[a]phenazine-1,2-dioxide and 2‑ethoxycarbonyl-3-methyl-quinoxaline-1,4-dioxide. Insofar as in the living cell, besides the electron-transfer reaction, the reaction of proton transfer occurs, it seemed expedient to elucidate the effect of protonation reactions on the mechanism of action of prodrugs. This study is aimed at elucidation of the mechanism of deoxygenation of the mentioned compounds under the model conditions, i.e., in the aprotic medium (DMF) and in the presence of proton donors (CH3COOH).
EXPERIMENTAL
Benzo[a]phenazine-7,12-dioxide was synthesized by the method of [14]. The physicochemical characteristics of this compound are analogous to those known from the literature [15]. 2-Ethoxycarbonyl-3-methyl-1,4-dioxide was synthesized from benzofuroxane and acetoacetic ester [16] and identified according to the mass-spectrometry data, 1Н and 13С NMR spectra by comparing with the known data [17].
Chronoamperograms and cyclic voltammograms were obtained by means of potentiostats IPC “Pro” MF (Ekoniks, Russia) and Р-4 (Elins, Russia). As the supporting electrolyte, we used solutions of perchlorate or tetrafluoroborate of teterabutyl ammonium (Acros Organics) in DMF (“extra dry”). Polarization curves were recorded according to the three-electrode circuit. As the working electrode, we used a glassy carbon electrode (d = 1.7 mm), the counter electrode represented a helix of platinum wire; as the reference electrode we used a saturated calomel electrode (SCE) which was connected with the working solution by a bridge containing a porous ceramic diaphragm and filled with supporting electrolyte solution. To remove oxygen from the working solution, we passed high-purity argon through it before recording each CA or CV curve. During experiments, argon was passed through the space above the solution surface in order to avoid its contact with air. The working electrode surface was polished before measuring each curve. The ECP measurements were carried out by a potentiostat switched according to the three-electrode circuit. The working electrode was made of graphite with the surface area of 4 cm2, the counter electrode was made of platinum with the surface area of 2 cm2, the reference electrode was a SCE. The cathodic and anodic compartments were separated by a glass membrane. The electrolysis was carried out at the constant potential in the first ER stage (–0.8 V) for compound 1 or at the potential in the stage that appeared in the presence of CH3COOH at –1.0 V until the complete exhaustion of depolarizer. The content of the starting compound was monitored by measuring CV curves. At the end of electrolysis, the solution was diluted by a factor of 3–5 with water, the precipitate was filtered and dried. The extracted products were analyzed by 1Н NMR spectroscopy and mass spectrometry.
ReSULTS AND DISCUSSION
As follows from the Introduction, the antitumoral activity of “bioreductive agents” is directly connected with the reactivity of products of their one-electron reduction. Hence, attention was paid to the electrochemical and chemical reactions that occurred at the potentials of the first ER stage of model compounds 1 and 2. In the beginning, we considered the electrochemical behavior of these compounds in an nonaqueous solvent, i.e., under the conditions where they formed sufficiently stable radical anions (RA) in the first stage. Table 1 shows the results of electrochemical studies in aprotic medium.
The first cathodic peak in CV curves of compound 1 is reversible (Fig. 1). The potential difference between the cathodic and the corresponding anodic peaks ΔEp = Epc – Epa is equal to 63 mV, which is close to the theoretical value (57 mV) for reversible reactions. The current in the first cathodic peak for compound 1 is equal to the current of one-electron oxidation of ferrocene under the same conditions (nCV = 0.81). According to CA measurements, the limiting ER current for compound 1 in the first stage corresponds to the transfer of one electron per molecule of compound 1 (nCA = 0.99). The ratio of anodic and cathodic currents in the CV of the first ER stage of compound 1 ipa/ipc is close to unity at the potential scan rate v ≥100 mV s–1.

CV curves of a solution of 5.0 mM of compound 1 in DMF (0.1 М Bu4NClO4) at 100 mV s–1 and different reversal potentials.
The value of Ipc varies linearly with the concentration of compound 1 in the interval from 3.9 to 18.8 mM and also with the square root of the potential scan rate \(({{v}^{{1{\text{/}}2}}})\) below 1 V s–1. These dependences pass through the origin, which points to the diffusion control in the first stage of ER of compound 1 to form RA 1 relatively stable in the aprotic medium.
It should be noted that we failed to find any literature data on the mechanism of ER for compound 1 in aprotic medium. Only the ER potentials for certain aromatic N,N′-dioxides of the close composition in DMSO are published, particularly, for benzo[a]phenazine-7,12-dioxides [13, 17]. The electrochemical behavior of phenazine-N,N′-dioxide was studied in more detail [18, 19]. Its ER produces RAs relatively stable in aprotic organic solvents and detectable by the CV and the EPR spectroscopy [18, 19]. The potential of the first ER stage of phenazine-N,N′-dioxide in DMF equal to –0.85 V [19] differs not so much from the value we determined for compound 1 (–0.91 V).
Under the conditions of several cycles in the interval from 0 to –1.2 V, in each subsequent CV curve the current of the first cathodic peak and the corresponding anodic peak remained approximately the same as in the first scan; however, near –1 V a relatively small new cathodic peak appeared which, as will be shown below, coincides as regards its potential with the ER peak of benzophenazine-N-monoxide—the reduction product of compound 1 in the presence of proton donors. Based on this, we can assume that in the aprotic medium, the RA of compound 1 is protonated very slowly by trace amounts of proton-donating impurities in solution to form electroactive products.
In the second ER stage of compound 1 (Е2 = –1.50 V) (Fig. 1), the second electron is transferred to molecule 1 to form the corresponding dianion. This ER stage is irreversible, which is associated with the higher basicity of the dianion as compared with the RA. The dianion of compound 1 is rapidly protonated even in virtually anhydrous DMF, which results in the formation of electrochemically active intermediate products the oxidation peaks of which are observed in the reverse CV branch (Fig. 1). The ER peaks at –1.78 and ‒2.00 V are associated with the reduction of the frame of the heteroaromatic system of compound 1.
To elucidate the fraction of protons at the ER of compound 1, we added the known amount of CH3COOH and measured CV and CA curves. In the presence of acid, the CV peak of the RA 1 oxidation at –0.85 V decreased (Fig. 2).

CV curves of a solution of 2.2 mM compound 1 with addition of CH3COOH at \(v\) = 100 mV s–1. CH3COOH concentration, mM: (1) 0, (2) 0.94, (3) 1.9, (4) 2.8, (5) 4.4.
For the acid concentration lower than the concentration of compound 1, the peak of RA oxidation is still detected in the CV curve. The equivalent content of the proton donor leads to disappearance of this peak. For 1–2 equivalents of CH3COOH with respect to the initial concentration of compound 1, the cathodic current in the first ER stage under these conditions (‒0.81 V) remained virtually unchanged amounting to the value approximately equal the current of the first cathodic peak in the aprotic DMF solution. In other words, the presence of a proton donor does not lead to formation of electrochemically active products in the first ER stage of compound 1. However, when we add 1/4 equivalent of CH3COOH with respect to the initial concentration of compound 1, new peaks are observed in the CV curve: at Е = –0.81, –1.05, and –1.25 V. Their current increases with the increase in CH3COOH concentration in solution. In this case, the current of the peak observed at the potential of the second-electron transfer to the depolarizer molecule Е = –1.5 V decreases (Fig. 2, curves 1–3). This ER stage completely disappears from the CV curve at the equal concentrations of the proton donor and compound 1.
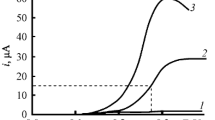
Figure 3 (curve 1) shows the changes in the current of peak at –0.81 V upon the addition of CH3COOH.

The number of electrons (n) involved in the ER of compounds (1) 1 and (2) 2 at the first-stage potential as a function of the relative (with respect to depolarizer) concentration of CH3COOH in DMF solution at \(v\) = 100 mV s–1.
The current is normalized (n = ipc/i0) to its value in the first ER stage of compound 1 in aprotic DMF (i0) that corresponds to the one-electron process; the concentration of CH3COOH in solution is normalized to the concentration of the studied compound. As seen from this figure, n remains close to 1 up to the 4-fold excess of acid. The analogous results were obtained by the CA method by processing the chronoamperograms recorded at the potentials of the preliminary stage for solutions of compound 1 with the addition of 1–4 equivalents of CH3COOH.
The above results allow us to assume that the peak that appears in the presence of proton donor at –0.81 V probably corresponds to the ER of a complex of compound 1 with acetic acid. It is known [20] that the formation of donor-acceptor complexes (molecular complexes) and even of solid salts with proton donors is typical of heteroaromatic N-oxides. Probably, the strong binding of compound 1 with CH3COOH by hydrogen bonds leads to a decrease in the energy of the lowest vacant orbital of molecular complex and, hence, to a decrease in its ER potential. The complexation of phenazine-N,N′-dioxide, which has the structure close to that compound 1, with acids (HClO4, H2SO4, CF3COOH) was proposed in [21] when studying its electroreduction in CH3CN.
To obtain experimental data on the possible reaction of compound 1 with acids, we collected UV spectra of DMF solutions of compound 1 containing CCl3COOH and CH3COOH.
As seen from the spectrum (Fig. 4), the absorption peak at λ = 472 nm decreases with the increase in the concentration of CCl3COOH, while the absorption intensity at 284–290 nm increases.

UV spectra of compound 1 in DMF in the presence of CH3COOH in the compound 1 : CH3COOH concentration ratio: (1) 1 : 0, (2) 1 : 1, (3) 1 : 2, (4) 1 : 3, (5) 1 : 4, (6) 1 : 5. Arrows show the direction of absorbance variation with the increase in the CH3COOH concentration.
In the case of CH3COOH, the effects in spectra are qualitatively the same but far less pronounced probably because CH3COOH is a weaker acid as compared with CCl3COOH. This character of variation of the UV spectra in the presence of a proton donor were also observed earlier for phenazine-N,N'-dioxide in CH2Cl2 [22]. Based on the results obtained by UV spectroscopy of DMF solutions of compound 1, we can assume that the formation of a complex is due to donor–acceptor interaction of this compound with CCl3COOH, and also with CH3COOH. The totality of results obtained by the UV spectroscopy and CV methods allows us to assume that at the potentials of the first ER stage of compound 1 in the presence of CH3COOH, the main reaction is the reduction of the complex formed by the original compound with the proton donor:

The competitive reaction of protonation of the RA of compound 1 does not produce any electroactive products at an insignificant excess of CH3COOH. Benzophenazine-N-monoxide which forms in this stage under these conditions is reduced reversibly at ‒1.05 V. When the CH3COOH concentration exceeds its equivalent value with respect to compound 1, a peak (Fig. 2, curves 4 and 5) appears at Е = –0.96 V which corresponds apparently to the ER of the complex of benzophenazine-N-monoxide with CH3COOH.

In other words, the deoxygenation of compound 1 proceeds consecutively at different potentials first for one N–O group and then for the other.
To reveal the nature of peaks that appear in CV curves of ER of compound 1 in the presence of low concentrations of proton donor, we carried out the electrolysis at the controlled potential of the first and second ER stages of compound 1 in the presence of СН3СООН. The electrolysis of solution containing 10 mM of compound 1 and the equivalent amount of СН3СООН was carried out at –0.8 V. After 1 F of electricity per initial concentration of compound 1 was passed, the current of the peak at –0.91 V virtually disappeared from the curve measured after the electrolysis (Fig. 5).

CV curves of compound 1 (10.7 mM) in the presence of CH3COOH (10 mM) (1) before electrolysis; (2) solution after the electrolysis (1 F); (3) CV curve of compound 1 in DMF.
Moreover, a reversible ER peak appeared at –1.05 V. Its current is close to that of the original compound 1. According to HRMS (high-resolution mass spectroscopy) data, the molar mass of the main ER product of compound 1 extracted after the electrolysis was equal to 247.06 g/mol (+М (Н)), and its 1Н NMR spectrum corresponded to the structure of benzo[a]phenazine-N-monoxide. These facts allow us to state that under the conditions described here, the ER of compound 1 produces exactly monoxide of benzo[a]phenazine.
As was mentioned above, in the presence of more than one equivalent of СН3СООН the CV curves demonstrate yet another reversible peak at –1.25 V (Fig. 2). Assuming that at this potential, it is the product of complete deoxigenation, i.e., benzo[a]phenazine, that is reduced, we carried out an ECP experiment at Е = –1.0 V. According to Fig. 6, after the exhaustive electrolysis under these conditions, the solution contained the ER product of compound 1 which demonstrated two cathodic peaks at –1.28 and –1.98 V, i.e., close to the potentials of two ER peaks of phenazine in DMF (–1.2 and –2.01 V) [23]. The mass spectrum and the 1Н NMR spectrum of the main ER product of compound 1 extracted after the electrolysis corresponded to characteristics of benzo[a]phenazine. Based on these data, we can state that the latter is the main ER product of compound 1 at the potential of –1.0 V under these conditions. According to the CV curve (Fig. 6), the concentration of benzo[a]phenazine in the catholyte after electrolysis is virtually equal to the concentration of the original compound before the electrolysis. Thus, the electrolysis produces benzo[a]phenazine in quantitative yield.

CV curves in (1) DMF solution of 10 mM of compound 1; (2, 3) the same solution with addition of 20 mM acetic acid (2) before the electrolysis and (3) after the electrolysis at Е = –1.0 V. Curve 4 —ER of benzo[a]phenazine (2.9 mM) extracted after the electrolysis from the catholyte.
The results of studying the ER of compound 1 in DMF in the presence of СН3СООН showed that the predominant direction of its transformation on the electrode is the ER of the complex of dioxide 1 with the proton donor. The homolytic cleavage of the N‒OH bond in the radical formed at the ER of this complex leads to the quantitative formation of mono-N-oxide and abstraction of an ·OH species. Mono-N-oxide also forms a complex with the proton donor added to solution and is reduced to benzo[a]phenazine forming the equivalent amount of active radicals •OH.
To reveal similarity or differences in the ER mechanisms of derivatives of benzophenazines and quinoxalines, we studied the ER mechanism of compound 2. Figure 7 shows the CV curves of compound 2 in DMF and, for a comparison, the ER curves of compound 1 (Fig. 1).

CV curves of compound 2 (4.6 mM) in DMF (0.1 М Bu4NBF4) on a GC electrode at different reversal potentials at \(v\) = 100 mV s–1.
The electrochemical behavior of compound 2 in the first stage in the aprotic medium is similar to that of compound 1 (Table 1). The current in the first ER peak of compound 2 changes linearly with both the concentration of depolarizer and \({{v}^{{1{\text{/}}2}}},\) which points to the formation of a relatively stable RA of compound 2. The subsequent ER stage in the CV of compound 2 in anhydrous aprotic DMF represents the sum of two peaks with close potentials in the vicinity of –1.65 and –1.76 V. Moreover, when the scan direction is reversed at –1.85 V, the reverse scan contains no peaks of deoxygenation products of dianion 2 (Fig. 7). In other words, in aprotic medium, the main target of protonation of dianion 2 is probably not the N–O bond as in ER of compound 1, but the heneroaromatic ring of molecule 2. The protonation of dianion of compound 2 produces a monoanion capable of reduction at –1.76 V and oxidation at –0.30 V in the reverse curve (Fig. 7).
In the presence of СН3СООН, these peaks in CV curves of compound 2 disappear (Fig. 8) but in this case, as for compound 1, the ER peaks of deoxygenation products of compound 2 appear and grow.

CV curves of compound 2 (4.6 mM) in DMF in the presence of СН3СООН, \({{c}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{COOH}}}}}\), mM = (1) 0, (2) 0.93, (3) 1.90, (4) 2.86, (5) 3.85, (6) 4.86 at \(v\) = 100 mV s–1.
According to Fig. 8, as in the case of ER of compound 1, the addition of СН3СООН leads to the appearance of the preliminary stage in CV curves of compound 2. The potential of this stage is –1.0 V. The peak current increases with the increase in the acid concentration to the greater extent as compared with compound 1 (compare with Fig. 3). Probably, the radical formed at the potential of this preliminary stage from the complex of compound 2 with СН3СООН is partly reduced on the electrode.

The RA of compound 2 is formed at the potential 200 mV more negative than the RA of compound 1; hence, bearing in mind the data of [24] on the correlation between the basicity of RA of aromatic compounds and the potential of their formation, we can state that the RA 2 enters into protonation reactions at the higher rate than the RA of compound 1.
The ER of the protonated RA of compound 2 leads probably to hydrogenation of the aromatic ring in molecule 2 because the electron density of an unpaired electron is distributed with the higher probability over the heteroaromatic part of its quinoxaline frame [25]. This is confirmed by the fact that the oxidation of the product of one-electron reduction of quinoxaline dioxides by oxygen proceeds not always reversibly but can produce the corresponding hydroxy derivative [26].
CONCLUSIONS
The experimental data obtained here suggest that in the aprotic medium, the formed radical anions of N,N′-dioxides of compounds 1 and 2 are stable. Deoxygenation of these compounds proceeds only in the presence of proton donors as a result of oxidation of N–O groups in two consecutive one-electron stages. Both stages are accompanied by the abstraction of radical •ОН to form mono-N-oxide of the corresponding derivative in the first stage and benzophenazine or quinoxaline in the second stage. According to electrochemical studies, the products can be formed by two routes. In the first route, it is the complex of depolarizer with the molecule of proton donor that is reduced (Scheme (1)). This mechanism is more probable for compound 1. The second route of transformation is the protonation of the RA of dioxide followed by decomposition of the radical (Scheme 2). If the radical Ar(N–O)2H• decomposes at a high rate, then both reactions generate active OH· radical in virtually quantitative yields. For the ER of compound 2, the Аr(N–O)2H• radical has time enough to be partly reduced on the electrode, which manifests itself by the increase in the emerging prepeak above the level of one-electron reduction; and, hence, the number of active ·OH radicals decreases. Thus, to characterize the activity of N,N'-dioxides in the reactions of generation of active oxygen-containing species, one must take account of the totality of main redox reactions and accompanying chemical reactions.
REFERENCES
Da Cunha, J., Lavaggi, M.L., Abasolo, M.I., Cerecetto, H., and Gonzalez M., 2D- and 3D-quantitative structure-activity relationship studies for a series of phenazine N,N'-dioxide as antitumour agents, Chem. Biol. Drug Des., 2011, vol. 78, p. 960.
Pachon, O. G., Azqueta, A., Lavaggi, M.L., Lopez de Cerain, A., Creppy, E., Collins, A., Cerecetto H., Gonzalez, M., Centelles, J.J., and Cascante, M., Antitumoral effect of phenazine N5,N10-dioxide derivatives on caco-2 cells, Chem. Res. Toxicol., 2008, vol. 21, p. 1578.
Gonda, M., Nieves, M., Nunes, E., Lopez de Cerain, A., Monge, A., Lavaggi, M. L., Gonzalez, M., and Cerecetto, H., Phenazine N,N0-dioxide scaffold as selective hypoxic cytotoxin pharmacophore. Structural modifications looking for further DNA topoisomerase II-inhibition activity, Med. Chem. Commun., 2013, no. 4, p. 595.
Lavaggi, M.L., Cabrera, M., Pintos, C., Arredondo, C., Pachon, G., Rodrıguez, J., Raymondo, S., Pacheco, J.P., Cascante, M., Olea-Azar, C., Lopez de Cerain, A., Monge, A., Cerecetto, H., and Gonzalez, M., Novel phenazine 5,10-dioxides release •OH in simulated hypoxia and induce reduction of tumour volume in vivo, ISRN Pharmacol., 2011, Article ID 314209. https://doi.org/10.5402/2011/314209
Chowdhury, G., Sarkar, U., Pullen, S., Wilson, W.R, Rajapakse, A., Fuchs-Knotts, T., and Gates, K.S., DNA strand cleavage by the phenazine di-N-oxide natural product myxin under both aerobic and anaerobic conditions, Chem. Res. Toxicol., 2012, vol. 25, p. 197. https://doi.org/10.1021/tx2004213
Fuchs, T., Chowdhury, G., Barnes, Ch.L., and Gates, K.S., 3-Amino-1,2,4-benzotriazine 4-oxide: characterization of a new metabolite arising from bioreductive processing of the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine), J. Org. Chem., 2001, vol. 66, p. 107.
Anderson, R.F., Shinde, S.S., Hay, M.P., Gamage, S.A., and Denny, W.A., Activation of 3-amino-1,2,4-benzotriazine 1,4-dioxide antitumor agents to oxidizing species following their one-electron reduction, J. Am. Chem. Soc., 2003, vol. 125, p. 748.
Yin, J., Glaser, R., and Gates, K.S., On the reaction mechanism of tirapazamine reduction chemistry: Unimolecular N−OH homolysis, stepwise dehydration, or triazene ring-opening, Chem. Res. Toxicol., 2012, vol. 25, p. 634. https://doi.org/10.1021/tx200546u
Shen, X., Laber, Ch.H., Sarkar, U., Galazzi, F., Johnson, K.M., Mahieu, N.G., Hillebrand, R., Fuchs-Knotts, T., Barnes, Ch.L., Baker, G.A., and Gates, K.S., Exploiting the inherent photophysical properties of the major tirapazamine metabolite in the development of profluorescent substrates for enzymes that catalyze the bioreductive activation of hypoxia-selective anticancer prodrugs, J. Org. Chem., 2018, vol. 83, no. 6, p. 3126. https://doi.org/10.1021/acs.joc.7b03035
Cimmino, A., Evidente, A., Mathieu, V., Andolfi, A., Lefranc, F., Kornienko, A., and Kiss, R., Phenazines and cancer, Nat. Prod. Rep., 2012, vol. 29, p. 487. https://doi.org/10.1039/c2np00079b
Cerecetto, H., Gonzalez, M., Lavaggi, M.L., Azqueta, A., Lopez de Cerain, A., and Monge, A., Phenazine 5,10-dioxide derivatives as hypoxic selective cytotoxins, J. Med. Chem., 2005, vol. 48, p. 21.
Hay, M.P., Gamage, S.A., Kovacs, M.S., Pruijn, F.B., Anderson, R.F., Patterson, A.V., Wilson, W.R., Brown, J.M., and Denny, W.A., Structure-activity relationships of 1,2,4-benzotriazine 1,4-dioxides as hypoxia-selective analogues of tirapazamine, J. Med. Chem., 2003, vol. 46, p. 169.
Lavaggi, M.L., Nieves, M., Cabrera, M., Olea-Azar, C., Lopez de Cerain, A., Monge, A., Cerecetto, H., and Gonzalez, M., Structural modifications on the phenazine N,N′-dioxide-scaffold looking for new selective hypoxic cytotoxins, Eur. J. Med. Chem., 2010, vol. 45, p. 5362.
Abu El-Haj, M. J., Dominy, B. W., Johnston, J.D., Haddadin, M.J., and Issidorides, C.H., A New route to phenazine 5,10-dioxides and related compounds, J. Org. Chem., 1978, vol. 7, no. 4, p. 589.
Lavaggi, M.L., Cabrera, M., Aravena, M. de los A., Olea-Azar, C., Lypez de Cerain, A., Monge, A., Pachyn, G., Cascante, M., Bruno, A.M., Pietra-santa, L.I., Gonzalez, M., and Cerecetto, H., Study of benzo[a]phenazine 7,12-dioxide as selective hypoxic cytotoxin-scaffold. Identification of aerobic-antitumoral activity through DNA fragmentation, Bioorg. Med. Chem., 2010, vol. 18, p. 4433.
El-Gogary, S.R., Waly, M.A., Ibrahim, I.T., and El-Sepelgy, O.Z., Synthesis and UV absorption of new conjugated quinoxaline1,4-dioxide derivatives anticipated as tumor imaging and cytotoxic agents, Monatsh. Chem., 2010, vol. 141, p. 1253. https://doi.org/10.1007/s00706-010-0386-1
Romeiro, N.C., Aguirre, G., Hernandez, P., Gonzalez, M., Cerecetto, H., Aldana, I., Perez-Silanes, S., Monge, A., Barreiro, E.J., and Lima, L.M., Synthesis, trypanocidal activity and docking studies of novel quinoxaline-N-acylhydrazones, designed as cruzain inhibitors candidates, Bioorg. Med. Chem. 2009, vol. 17, p. 641.
Kulakovskaya, S.I., Krivenko, A.G., Komarova, N.S., Kulikov, A.V., and Shestakov, A.F., Electrochemical and ESR study of the mechanism of oxidation of phenazine-di-N-oxide in the presence of cyclohexanol on glassy carbon and single-walled carbon nanotube electrodes, Russ. J. Electrochem., 2014, vol. 50, p. 1.
Miyazaki, H., Matsuhisa, Y., and Kubota, T., Cyclic voltammetry of aromatic amine N-oxides in nonaqueous solvents and the stability of the free radicals produced, Bull. Chem. Soc. Jpn., 1981, vol. 54, p. 3850.
Ryzhakov, A.V., Alekseeva, S.A., and Rodina, L.L., New trends in the chemistry of molecular complexes of heteroaromatic N-oxides, Vestn. St.-Petersburg Univ., Ser. 4, 2009, no. 1, p. 67.
Koldasheva, E.M., Shestakov, A.F., Geletii, Yu.V., and Shilov, A.E., Formation and redox properties of a complex of phenazine di-N-oxide with a proton, Bull. Russ. Acad. Sci. Chem., 1992, vol. 41, no. 4, p. 655.
Koldasheva, E.M., Strelets, V.V., Tse, Y.K., Geletii, Yu.V., and Shestakov, A.F., Phenazine di-N-oxide radical cation and its reaction with hydrocarbons, Russ. Chem. Bull., 1996, vol. 45, no. 8, p. 1889.
Tabner J. and Yandle J.R., A Correlation of half-wave reduction potentials with theoretical calculations for some nitrogen-containing heteromolecules in dimethylformamide, J. Chem. Soc. A, 1968, p. 381.
Mendkovich, A.S., Syroeshkin, M.A., Mikhalchenko, L.V., Mikhailov, M.N., Rusakov, A.I., and Gul’tyai, V.P., Integrated study of the dinitrobenzene electroreduction mechanism by electroanalytical and computational methods, Int. J. Electrochem., vol. 2011, Article ID 346043. https://doi.org/10.4061/2011/346043
Chowdhury, G., Kotandeniya, D., Daniels, J.S., Barnes, Ch.L., and Gates, K.S., Enzyme-activated, hypoxia-selective DNA damage by 3-amino-2-quinoxalinecarbonitrile 1,4-di-N-oxide, Chem. Res. Toxicol., 2004, vol.17, no. 11, p. 1399. https://doi.org/10.1021/tx049836w
Ganley, B., Chowdhury, G., Bhansali, J., Daniels, J.S., and Gates K.S., Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide, Bioorg. Med. Chem., 2001, vol. 9, p. 2395.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare the absence of conflict of interests.
Additional information
Translated by T. Safonova
Published on the basis of materials of the XIX All-Russian Conference “Electrochemistry of Organic Compounds” (EKHOS-2018) (with international participation), Novocherkassk, 2018.
Rights and permissions
About this article

Cite this article
Mikhal’chenko, L.V., Nasybullina, D.V., Leonova, M.Y. et al. Electroreduction of Derivatives of N,N'-Dioxides of Phenazine and Quinoxaline in Nonaqueous Media and in the Presence of Proton Donors of Medium Strength. Russ J Electrochem 56, 388–395 (2020). https://doi.org/10.1134/S1023193520040102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1023193520040102