
Abstract
At present, transcriptomics is one of the fastest developing fields of molecular biology, which allows to obtain detailed information about the functional activity of the genome in both normal and pathological conditions. We used modern transcriptomic technologies to comprehensively characterize the whole genome gene expression profile of human placental syncytiotrophoblast cells (STB) in physiological pregnancy and preeclampsia (PE). As a result of our analysis, we identified 26 differentially expressed genes (DEGs) in the STB cells between healthy and diseased states. The cluster of DEGs contains not only well-known candidate genes identified earlier in many foreign whole genome studies of the placenta (for example, LEP, INHBA, and FLT1) but also new genes (AC098613.1, AC087857.1, FCRLB, TENM4, PTP4A1P7, LINC01225, etc.) that can be considered as new biological markers of PE and are of interest for further study. Functional enrichment annotation indicated that most of the DEGs were implicated in the signaling pathways of regulation of hormonal secretion, MAPK cascade, ERK1 and ERK2 cascade, positive regulation of cell adhesion, and proliferation of endothelial cells. These processes may be associated with the development of PE at the level of STB cells. Additionally, we revealed that alternative splicing of the FLT1 gene indicates the important role of this RNA processing mechanism in the pathogenetics of PE due to a significant increase in the transcriptional diversity of genes in STB cells. The expression level of the transcript encoding the protein isoform FLT-1 e15a was significantly increased in patients with PE compared to the control group. This study expands understanding of the molecular mechanisms involved in PE and can serve as a basis for development of preventive, prognostic, and therapeutic strategies in the field of personalized obstetrics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The personalized focus of modern medicine requires innovative approaches to the study of the molecular mechanisms of diseases, as well as the development of effective methods of targeted therapy based on experimental results obtained using omics technologies. In this context, the analysis of the transcriptome of various tissues and cells is of considerable interest to researchers, since the disruption of the expression activity of genes is involved in the pathogenesis of numerous diseases. Today, transcriptomics is one of the fastest growing areas of molecular biology, providing detailed information about the functional activity of the human genome. The transcriptome reflects the gene expression profile at a given time and, unlike the genome, is dynamic and actively changes depending on external factors and the needs of surrounding tissues and cells, which makes it a convenient tool for studying the mechanisms of implementation of genetic information in the process of adaptation of cells to changing environmental conditions, as well as the ways of their interaction in normal and pathological conditions. In view of this, transcriptomic technologies have found wide application in various areas of predictive medicine, for example, in the development of diagnostic methods and strategies for predicting and preventing various diseases, including obstetric pathology, where the placenta is the main object of study. It should be noted that the study of molecular processes occurring in placental tissue and associated with the transcriptional regulation of gene expression is considered today the most promising area of research for revealing the pathophysiology of complicated pregnancy, in particular, such a serious disease as preeclampsia [1].
Molecular mechanisms of development of preeclampsia (PE) have been the subject of fundamental and applied research in the field of medicine and biology for more than 40 years. However, despite some progress in this area, attempts to form a unified theory of the etiopathogenesis of PE have not yet been successful. Over the past decade, using methods of biochemistry and molecular genetics, it has been possible to identify individual predictive markers for the development of PE (for example, PIGF and sFLT1), data on the prognostic significance of which vary depending on the studies [2, 3]. Today, only one etiological point in the development of this gestational complication is not discussed—the presence of pregnancy and impaired formation of the spiral arteries of the placenta. Presumably, a variety of triggers can play a significant role in the occurrence of PE, which cause the inclusion of many pathogenetic links in the development of this severe pathology, which ultimately manifests itself in multiple organ failure and the involvement of various tissues and cell types in the pathological process [4]. The improvement of high-throughput methods of transcriptomics has made it possible to analyze the genome-wide profile of mRNA in individual human cells, which makes it possible to identify new biomarkers and functionally significant signal transduction pathways, as well as networks of intermolecular and intercellular interactions, and thereby expand the understanding of the molecular mechanisms of this severe obstetric disease, as well as develop new personalized strategies for therapeutic and preventive measures.
It is generally accepted that the main structural element and regulator of placental functions is syncytiotrophoblast (STB), lining the entire surface area of the placental villi. Owing to direct contact with maternal blood, STB performs many functions of the placenta, including gas exchange, nutrient transport, fetal protection, and the secretion of a number of hormones and mediators, which subsequently enter the maternal bloodstream [5]. Moreover, any disturbances in the functioning of STB, starting with the processes of invasion, can cause the development of subsequent complications of pregnancy, including PE. It has been shown that mediators released from placental tissue into the maternal blood are associated with clinical symptoms of PE such as high blood pressure and protein in the urine. Moreover, it was found that under oxidative stress, STB releases a number of complex substances into the maternal bloodstream, including pro-inflammatory cytokines, exosomes, and antiangiogenic factors, which can disrupt the function of the maternal endothelium and lead to a systemic inflammatory response and the formation of a pathological course of pregnancy [5, 6].
It is worth noting that, to date, numerous foreign and isolated domestic studies devoted both to characterizing the gene expression profile during physiological pregnancy and to searching for transcriptome patterns of blood and placenta leukocytes associated with various gestational complications have been conducted [1, 6–11]. However, it is still unclear what changes occur in the gene expression profiles of different types of placental tissue cells under conditions of normal and pathological pregnancy. In the presented work, we used modern transcriptomic technologies to comprehensively characterize the genome-wide gene expression profile of human placenta STB cells during physiological and complicated PE pregnancies.
MATERIALS AND METHODS
The conducting of this study was approved by the Biomedical Ethics Committee of the Research Institute of Medical Genetics of the Tomsk National Research Medical Center. The material for the study is represented by biopsy samples of the maternal part of the placenta of women with a physiological course of pregnancy (N = 8) and severe preeclampsia (N = 8). The collection of biopsy specimens was carried out according to the standard method [12] by obstetricians and gynecologists at the facilities of the Yevtushenko Regional Perinatal Center, Tomsk. The resulting material was immediately washed with saline and placed in cryovials, which were then stored in liquid nitrogen to minimize RNA degradation. In all patients, placental tissue sections were characterized histologically with hematoxylin-eosin staining. Laser microdissection to obtain STB cells was carried out on PALM equipment (Carl Zeiss, Germany) with the technology of automated fragment capture (laser capture microdissection).
To isolate total RNA, the Single Cell RNA Purification Kit (Norgen, United States) was used. RNA concentration and quality were assessed using an Agilent 2100 Bioanalyzer. Library preparation was carried out according to the SMARTer Stranded Total RNA-Seq Kit v2 protocol (Takara, United States). Mass parallel sequencing was performed on a Next-seq 2000 instrument (Illumina). Alignment to the reference genome (hg38) was performed using the STAR program. Annotation of transcription active regions obtained in this work as a result of sequencing of STB cells was carried out in the R software environment (http://www.r-progect.org) using the gencode.v39 database. Data filtering and normalization were carried out in the edgeR package of the R statistical computing environment according to standard parameters. Statistical analysis of differential expression was also performed using the edgeR package. The threshold for filtering differentially expressed RNAs was |log2FC| > 1 and FDR < 0.05 as optimal for minimizing errors of the second type, since the methods of subsequent statistical and integrative analyses are resistant to errors of the first type [13].
Functional analysis of a cluster of differentially expressed genes (DEG) was carried out using a web tool in the Molecular Signatures Database (MSigDB) resource of the GSEA software and the GeneMANIA database [14, 15]. Analysis of alternative splicing events was performed using the MAJIQ package [16], which makes it possible to predict and quantify splicing events on the basis of whole-genome RNA sequencing data, using BAM and GFF3 input files. Reconstruction of alternative isoform sequences was performed using the Illumina DRAGEN (Dynamic Read Analysis for GENomics) Bio-IT Platform. Experimental studies were carried out at the Center for Shared Use of Research Equipment Medical Genomics of the Research Institute of Medical Genetics of the Tomsk National Research Medical Center.
RESULTS AND DISCUSSION
To establish the molecular mechanisms that may underlie the formation of preeclampsia at the level of dysfunction of the placental tissue villi, a transcriptomic analysis of STB cells from patients with PE and women with physiological pregnancy was carried out using RNA-seq technology.
As part of this work, 16 STB cell transcriptomes were obtained from placental tissue samples of eight patients with PE and eight women from the control group. At least 86% of the sequence reads were uniquely aligned to the human genome, and the uniformity of read mapping across the transcripts of all samples was good. After alignment to the human genome, the average coverage per sample was 43 million reads. A total of 22 625 transcripts were included in the analysis, for which the read count (CPM) was at least five for 10% or more of the samples. Annotation of transcriptional active regions obtained in this work as a result of sequencing of STB cells was carried out in the R software environment using the gencode.v39 database (see Table 1; averaged data for all analyzed samples are given). Most of the analyzed transcripts (66.5%) correspond to protein-coding regions of the human genome (15 046). The remaining cluster is dominated by long noncoding RNAs and processed pseudogenes.
Analysis of Differential Expression of Syncytiotrophoblast Genes
Differential expression of STB genes was determined using the edgeR package, which calculates the variance of the expression score for each gene. Genes with differences in the absolute value of expression on the logarithmic scale |log2FC| > 2 and a significance level adjusted for multiple comparisons of less than 0.05 were considered differentially expressed (Fig. 1).

Volcano plot showing the dependence of statistical significance (FDR level) on the fold difference in gene expression between groups. Top differentially expressed genes in PE and physiological pregnancy (|log2FC| > 2 and FDR < 0.05) are marked with red dots.
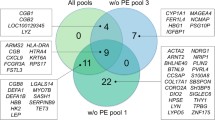
As a result of our analysis, 26 genes were identified whose expression differs statistically significantly (FDR < 0.05; |log2FC| > 1) in STB cells of women with PE and a physiological course of pregnancy (Fig. 2). And only for two genes, IGHG1 and TUBG2, encoding the constant region of the heavy chain of gamma globulin 1 and the gamma tubulin chain, respectively, was a decrease in transcriptional activity found in the group of patients compared to the control sample, while overexpression in patients with PE was shown for 24 genes, which is more than 92% of all identified DEGs.

Heat map of DEGs obtained from a comparative analysis of full transcriptomic profiles of STB cells in groups with physiological pregnancy (belonging to this group is indicated as C in the figure) and PE (samples in this case are labeled as PE).
The cluster of DEGs whose expression is increased in STB cells during PE includes not only known candidate genes previously identified in many genome-wide studies of placental gene expression profiles in this pathology (for example, LEP, FLT1, INHBA) but also new potential genetic markers (PTP4A1P7, AC087857.1, SLC46A3, RPS18P9, STAMBPL1, SULT1C2, AC073410.2, ASRGL1, TP53TG1, etc.), the connection of which with the development of PE was established for the first time in this work. It is also worth noting the large number of noncoding RNAs in this DEG cluster, the functional significance of which in the mechanisms of the pathological course of pregnancy has yet to be characterized. Table 2 presents data on the most significant DEGs (FDR < 0.01).
In order to assess the potential biological significance of the identified DEGs in the molecular mechanisms of PE, we carried out functional annotation and analysis of network interactions of the identified genes using the GSEA (Gene Set Enrichment Analysis) computational method and the GeneMANIA database. In the presented work, when performing functional annotation in the Molecular Signatures Database (MSigDB) resource of the GSEA software, categories with FDR (false discovery rate) <0.05 were taken into account (in this case, FDR is the probability of a nonrandom entry of a group of genes into a category adjusted for multiple comparisons). We identified eight categories including nine genes out of 26 studied. It should be noted that most of these genes are simultaneously involved in the implementation of several gene ontologies and molecular processes (for example, loci FGB, FGG, LEP, INHBA, TPBG, and FLT1 are included in at least three or more of the identified categories). The most significant biological pathways, processes, and molecular functions enriched in DEG data (FDR < 0.005) are associated with the regulation of hormonal secretion, regulation of the MAPK cascade, and positive regulation of processes in a multicellular organism (Table 3).
It is important to note that, among the DEGs, genes associated with the regulation of the MAPK cascade are overrepresented. Previous studies have shown that oxidative stress activates mitogen-activated protein kinase (MAPK) signaling and increases the expression of apoptosis receptors [17]. Activity of p38 MAPK was significantly increased in the placenta of patients with PE compared with women with normal pregnancies. Presumably, reactive oxygen species (ROS) may activate the p38 MAPK signaling pathway in placental tissue with subsequent overexpression of sFlt-1 and sEng in maternal serum and a systemic inflammatory response, causing endothelial dysfunction and clinical symptoms of PE [18]. It has been shown that inactivation of the Ras-MAPK signaling pathway in trophoblast cells is one of the main pathogenetic factors of idiopathic recurrent miscarriage owing to inhibition of the ability of trophoblast cells for proliferation and invasion [19].
The network of interactions between DEG products obtained using the GeneMANIA web resource confirms the relationships between genes identified in the study of biological pathways and processes. Analysis of this network, which includes 15 proteins encoded by the genes under study, indicates an important place in the molecular mechanisms of PE at the level of STB cells of protein–protein interactions of DEG products and co-expression of a number of genes (Fig. 3). Functional annotation of a cluster of DEGs connected by network interactions indicates a highly significant overrepresentation of these genes (FDR < 0.001) in such categories as endothelial and epithelial cell proliferation, regulation of endothelial cell proliferation, positive regulation of cell adhesion, and regulation of cytokine receptor binding, ERK1/ERK2 cascade, and intercellular adhesion, as well as negative regulation of the apoptosis process.

Network of protein–protein interactions of DEG products. The network of potential interactions was created using the GeneMANIA database (http://www.genemania.org). Accordingly, each colored line represents a specific type of functional association available in the literature, derived from different biological systems, and accepted by the GeneMANIA algorithm. Physical interaction—pink, co-expression—purple, common protein domain—yellow, co-localization—blue, common pathological pathway—light blue. The thicker the line, the closer the weight of association of the two given genes to each other.
The central place in this network with the largest number of interactions is occupied by genes FGB, FGG, LEP, and FLT1, encoding fibrinogen gamma and beta chains, leptin, and vascular endothelial growth factor receptor type 1, respectively. The products of some of these genes, according to the currently known data on their functional features, can play a leading role in the molecular mechanisms of the studied pathology.
Thus, modern studies demonstrate that an imbalance between pro- and antiangiogenic growth factors is of great importance in the processes of cytotrophoblast invasion in PE [3, 20]. Soluble antiangiogenic factor (sFlt-1) has been shown to be a product of gene FLT1; during the physiological course of pregnancy, it reduces the rate of cytotrophoblast invasion, while its concentration at the beginning of pregnancy is relatively low and begins to increase in the last trimester of pregnancy. S.A. Karumanchi et al. demonstrated significant overexpression of the isoform of gene FLT1 in the placental tissue, encoding sFlt-1 [20]. Along with this, immunohistochemistry methods revealed significant changes in the expression of VEGF, placental growth factor (PlGF), and antiangiogenic factor (Flt-1) in the placentas of women with PE. Mice with genome knockdown of these loci show impaired placental vasculogenesis and increased fetal mortality [21]. The results obtained in this work also indicate the key role of overexpression of gene FLT1 in the molecular mechanisms of PE at the level of STB cells, in connection with which we carried out a detailed analysis of the events of alternative splicing of this locus.
Analysis of Alternative Splicing of the FLT1 Gene
Analysis of alternative splicing (AS) events in the STB cells of the examined women was performed using the MAJIQ program [16]. This program identifies complex (LSV (local splice variation) events) and binary (seven “classic” types of events) AS events; annotated and novel, using RNA-Seq data, LSVs are defined and easily visualized as splits (multiple edges) in the splicing graph, where multiple edges either enter or originate from a single exon. In particular, previously defined “classical” AS events are mapped to special cases of binary graph splits (e.g., exon inclusion or skipping), while LSVs capture nonclassical binary splits and splits involving more than two junctions. Quantification of AS events under specific conditions is based on the marginal percentage splicing index (PSI, Ψ) from 0.05 to 0.95 for each element participating in the event.
In the control group for the gene FLT1, two annotated events were identified. One of them is complex LSV and includes an alternative last exon with a PSI level of 0.695 and intron retention with a PSI level of 0.293 (Fig. 4c); the constitutive exon–exon junction event, which has a low percentage of inclusion in the transcript and PSI = 0.0113, is indicated in red. The second event is a classic binary event and corresponds to the alternative last exon with PSI = 0.104, while the constitutive exon–exon event has PSI = 0.896 and is more likely to be included in the transcript (Fig. 4d).

Visualization of AS events for gene FLT1 in the control group using the MAJIQ VOILA program. (a) Intron-exon structure of the gene FLT1; (b) statistically significant events in the intron-exon structure; (c) annotated complex event; (d) annotated binary event.
In the group with preeclampsia for the gene FLT1, two annotated and two de novo events are identified. If we consider the group of annotated events, they have the same structure as in the control group, but a slightly different percentage splicing index. One of them is complex LSV and includes an alternative last exon with a PSI level of 0.702 and intron retention with a PSI level of 0.285 (Fig. 5d); the constitutive exon–exon junction event, which has a low percentage of inclusion in the transcript and PSI = 0.0131, is indicated in red. The second event is a classic binary event and corresponds to the alternative last exon with PSI = 0.338, while the constitutive exon–exon event has PSI = 0.662 and is more likely to be included in the transcript (Fig. 5e). Two events de novo are binary and correspond to intron retention between exons 6 and 7 (Fig. 5c) and between exons 37 and 38 (Fig. 5f). Both events have a low percentage splicing index of 0.0662 and 0.117, respectively.

Visualization of AS events for gene FLT1 in the preeclampsia group using the MAJIQ VOILA program. (a) Intron-exon structure of the gene FLT1; (b) statistically significant events in the intron-exon structure; (c) de novo binary event 1; (d) annotated complex event; (e) annotated binary event; (f) de novo binary event 2.
When assessing the level of expression of individual isoforms of the gene FLT1 in all 16 samples, the following transcripts were presented (Fig. 6): ENST00000282397.9 (encodes the FLT-1 protein), ENST00000541932.5 (sFLT-1 e15b), ENST0000061-5840.4 (sFLT-i13), ENST00000639477.1 (sFLT-e15a), and ENST00000540678.2, which does not have a protein product. According to the Mann–Whitney U test, the expression of transcripts ENST00000282397.9 (Z = ‒2.941, p = 0.0019) and ENST00000639477.1 (Z = ‒2.836, p = 0.0029) differs statistically significantly between the control group and the group with preeclampsia.

Variability of isoforms of gene FLT1 in STB cells.
It is noteworthy that the expression level of the transcript encoding the protein isoform FLT-1 e15a, which, according to the literature, is overexpressed in the blood and syncytiotrophoblast during PE [22], was significantly increased in patients with PE compared to the control group (thus, the average number of CPM was 88.21 in the control group and 390.15 in the preeclampsia group). In experiments in vitro in cell cultures, the sFlt-1e15a protein reduced endothelial cell migration and invasion and inhibited tubule formation in endothelial cells [22]. It is assumed that this isoform of FLT-1 can cause endothelial dysfunction and dysfunction of target organs affected by PE [23], which allows us to consider this option as promising in relation to the development of modern diagnostic methods and targeted therapy for this severe gestational disease.
Transcriptomic studies of multifactorial diseases are one of the priority areas of modern personalized medicine, including in the field of obstetrics. Despite significant progress in the study of the etiopathogenesis of the complicated course of pregnancy, many questions related to the molecular mechanisms of this condition remain open and require detailed study. Modern methods of transcriptomics provide a suitable methodological basis for implementing such studies. The main link in the pathogenesis of preeclampsia is a disruption of placentation with insufficient invasion of the trophoblast into the structures of the uterus and defective remodeling of the spiral arteries, leading to a decrease in blood supply and ischemia of the placenta. It has been shown that the main structural element and regulator of the functions of the formed placenta is syncytiotrophoblast, which lines the placental villi and is in direct contact with maternal blood. The presented work is the first full-transcriptomic study of syncytiotrophoblast cells from the human placenta in Russia. Twenty-six genes were identified whose expression differed statistically significantly in STB cells of women with PE and those with a physiological course of pregnancy. The DEG cluster contains not only known candidate genes previously identified in many foreign genome-wide studies of the placenta (for example, LEP, INHBA, and FLT1) but also new genes (AC098613.1, AC087857.1, FCRLB, TENM4, PTP4A1P7, LINC01225, etc.) which can be considered as new biological markers of PE and are of interest for further study. The results of the functional annotation of DEGs show that the development of PE at the level of STB may be associated with signaling pathways of regulation of hormonal secretion, MAPK cascade, ERK1 and ERK2 cascade, positive regulation of cell adhesion, proliferation of endothelial cells, etc. The results of the analysis of the AS gene FLT1 indicate an important role in the pathogenetics of PE of this RNA processing mechanism, which significantly increases the transcriptional diversity of PE candidate genes in STB cells.
Placental transcriptome profiling at the level of single cells and individual subpopulations of placental tissue cells, as well as integrative analysis with genomic, proteomic, and methylome data, represents a huge potential for expanding knowledge about the functioning of the placenta in both physiological and pathological pregnancy, for identifying the main causal mechanisms development of PE, and for predicting and improving maternal and perinatal outcomes in complicated pregnancy.
REFERENCES
Gong, S., Gaccioli, F., Dopierala, J., et al., The RNA landscape of the human placenta in health and disease, Nat. Commun., 2021, vol. 12, no. 1, p. 2639. https://doi.org/10.1038/s41467-021-22695-y
Dimitriadis, E., Rolnik, D.L., Zhou, W., et al., Pre-eclampsia, Nat. Rev. Dis. Primers, 2023, vol. 9, no. 1, p. 8. https://doi.org/10.1038/s41572-023-00417-6
Verlohren, S., Brennecke, S.P., Galindo, A., et al., Clinical interpretation and implementation of the sFlt-1/PlGF ratio in the prediction, diagnosis and management of preeclampsia, Pregnancy Hypertension, 2022, vol. 27, pp. 42—50. https://doi.org/10.1016/j.preghy.2021.12.003
Jena, M.K., Sharma, N.R., Petitt, M., et al., Pathogenesis of preeclampsia and therapeutic approaches targeting the placenta, Biomolecules, 2020, vol. 10, no. 6, p. 953. https://doi.org/10.3390/biom10060953
Redman, C.W.G., Staff, A.C., and Roberts, J.M., Syncytiotrophoblast stress in preeclampsia: the convergence point for multiple pathways, Am. J. Obstet. Gynecol., 2022, vol. 226, no. 2, pp. S907—S927. https://doi.org/10.1016/j.ajog.2020.09.047
Tsang, J.C.H., Vong, J.S., Ji, L., et al., Integrative single-cell and cell-free plasma RNA transcriptomics elucidates placental cellular dynamics, Proc. Natl. Acad. Sci. U.S.A., 2017, vol. 114, no. 37, pp. E7786—E7795. https://doi.org/10.1073/pnas.1710470114
Li, H., Huang, Q., Liu, Y., and Garmire, L.X., Single cell transcriptome research in human placenta, Reproduction (Cambridge, England), 2020, vol. 160, no. 6, p. R155. https://doi.org/10.1530/REP-20-0231
Benton, S.J., Leavey, K., Grynspan, D., et al., The clinical heterogeneity of preeclampsia is related to both placental gene expression and placental histopathology, Am. J. Obstet. Gynecol., 2018, vol. 219, no. 6, pp. 604.e1—604.e25. https://doi.org/10.1016/j.ajog.2018.09.036
Trifonova, E.A., Gabidulina, T.V., Ershov, N.I., et al., Analysis of the placental tissue transcriptome of normal and preeclampsia complicated pregnancies, Acta Nat., 2014, vol. 6, no. 2 (21), pp. 71—83. https://doi.org/10.32607/20758251-2014-6-2-71-83
Vashukova, E.S., Glotov, A.S., Fedotov, P.V., et al., Placental microRNA expression in pregnancies complicated by superimposed pre-eclampsia on chronic hypertension, Mol. Med. Rep., 2016, vol. 14, no. 1, pp. 22—32. https://doi.org/10.3892/mmr.2016.5268
Liu, S., Xie, X., Lei, H., et al., Identification of key circRNAs/lncRNAs/miRNAs/mRNAs and pathways in preeclampsia using bioinformatics analysis, Med. Sci. Monit.: Intern. Med. J. Exp. Clin. Res., 2019, vol. 25, p. 1679. https://doi.org/10.12659/MSM.912801
Robson, S.C., Simpson, H., Ball, E., et al., Punch biopsy of the human placental bed, Am. J. Obstet. Gynecol., 2002, vol. 187, no. 5, pp. 1349—1355. https://doi.org/10.1067/mob.2002.126866
Robinson, M.D., McCarthy, D.J., and Smyth, G.K., edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics, 2010, vol. 26, no. 1, pp. 139—140. https://doi.org/10.1093/bioinformatics/btp616
Liberzon, A., Subramanian, A., Pinchback, R., et al., Molecular signatures database (MSigDB) 3.0, Bioinformatics, 2011, vol. 27, no. 12, pp. 1739—1740. https://doi.org/10.1093/bioinformatics/btr260
Franz, M., Rodriguez, H., Lopes, C., et al., GeneMANIA update 2018, Nucleic Acids Res., 2018, vol. 46, no. W1, pp. W60—W64. https://doi.org/10.1093/nar/gky311
Vaquero-Garcia, J., Aicher, J.K., Jewell, S., et al., RNA splicing analysis using heterogeneous and large RNA-seq datasets, Nat. Commun., 2023, vol. 14, no. 1, p. 1230. https://doi.org/10.1038/s41467-023-36585-y
Wang, Z., Zhao, G., Zibrila, A.I., et al., Acetylcholine ameliorated hypoxia-induced oxidative stress and apoptosis in trophoblast cells via p38 MAPK/NF-κB pathway, Mol. Hum. Reprod., 2021, vol. 27, no. 8. https://doi.org/10.1093/molehr/gaab045
Guo, L., Liu, M., and Duan, T., Hydrogen suppresses oxidative stress by inhibiting the p38 MAPK signaling pathway in preeclampsia, Adv. Clin. Exp. Med., 2023, vol. 32, no. 3, pp. 357—367. https://doi.org/10.17219/acem/154623
Zhang, J., Liu, X., and Gao, Y., Abnormal H3K27 histone methylation of RASA1 gene leads to unexplained recurrent spontaneous abortion by regulating Ras-MAPK pathway in trophoblast cells, Mol. Biol. Rep., 2021, vol. 48, no. 6, pp. 5109—5119. https://doi.org/10.1007/s11033-021-06507-6
Karumanchi, S.A., Angiogenic factors in preeclampsia: from diagnosis to therapy, Hypertension, 2016, vol. 67, no. 6, pp. 1072—1079. https://doi.org/10.1161/HYPERTENSIONAHA.116.06421
Hiratsuka, S., Minowa, O., Kuno, J., et al., Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice, Proc. Natl. Acad. Sci. U.S.A., 1998, vol. 95, no. 16, pp. 9349—9354. https://doi.org/10.1073/pnas.95.16.9349
Palmer, K.R., Kaitu’u-Lino, T.U.J., Hastie, R., et al., Placental-specific sFLT-1 e15a protein is increased in preeclampsia, antagonizes vascular endothelial growth factor signaling, and has antiangiogenic activity, Hypertension, 2015, vol. 66, no. 6, pp. 1251—1259. https://doi.org/10.1161/HYPERTENSIONAHA.115.05883
Palmer, K., Assessing the circulating placental-specific anti-angiogenic protein sFLT-1 e15a in preeclampsia, Preeclampsia: Methods Protoc., 2018, pp. 27—37. https://doi.org/10.1007/978-1-4939-7498-6_3
Funding
This work was supported by ongoing institutional funding. No additional grants to carry out or direct this particular research were obtained.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research ethics committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed voluntary consent was obtained from each of the participants included in the study.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Trifonova, E.A., Babovskaya, A.A., Zarubin, A.A. et al. Transcriptomic Profiling of Placental Cells in Preeclampsia as an Effective Tool for Personalized Medicine. Russ J Genet 59, 1366–1377 (2023). https://doi.org/10.1134/S102279542312013X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S102279542312013X