
Abstract
Despite the development of exome and whole genome sequencing technologies and their routine use in the diagnosis of hereditary diseases, the efficiency of detection of pathogenic genetic variants for methods based on DNA analysis is less than 50%. One of the main reasons may be the inefficiency of these approaches in the search for genetic variants responsible for impaired pre-mRNA splicing. This review discusses the results of work on the search for splicing abnormalities in hereditary orphan diseases using RNA sequencing and the possibility of clinical application of this method.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
A distinctive feature of rare (orphan) diseases is the low frequency of occurrence in the population. However, the number of nosologies themselves is extremely large and increases every year. In the world, according to expert estimates, about 6172 rare diseases are known [1], most of them (71.9%) are based on a genetic nature [2]. The online database of Mendelian hereditary human diseases describes 5918 phenotypes of various hereditary nosologies with known molecular mechanisms for the development of a pathological condition and more than 3000 disease phenotypes without identified molecular mechanisms [3].
At present, the total number of patients with orphan diseases in the world is approximately 3.5–5.9% of the total population of the Earth, which corresponds to 263 to 446 million people [2]. At the same time, there are no such data for the Russian Federation at the moment. On the basis of the data of the average European and average world indicators of the prevalence of orphan diseases, as well as the population of the Russian Federation (146.9 million people as of 2018), it can be assumed that the potential number of patients with rare diseases in the Russian Federation can be from 5.1 to 8.6 million people.
Another feature of these diseases is the extremely high heterogeneity both in terms of the systems and organs they affect and in the degree of clinical manifestation. All this together leads to significant difficulties in diagnosing orphan diseases. For a patient, establishing a correct diagnosis over the course of several years may involve undergoing many diagnostic tests, as well as visiting a large number of specialists. As a result, some patients do not survive to be diagnosed. Given the heterogeneity of hereditary orphan diseases, one of the conditions for improving diagnostics is to expand the range of technologies used to search for known hereditary mutations and map new genes and genetic variants associated with the development of hereditary diseases.
Identification of etiologically significant mutations allows clarifying the molecular diagnosis in patients. This is necessary for medical genetic counseling, as it allows assessing the inheritance of pathogenetically significant mutations and makes it possible to identify carriers of mutations and family forms of diseases.
Making an accurate molecular diagnosis significantly improves the possibility of carrying out preventive measures. Thus, early diagnosis of hereditary metabolic diseases allows the use of replacement therapy, which in turn leads to the normalization of functions or a decrease in the severity of the pathological process. For some hereditary diseases, intrauterine treatment is possible (for example, with certain aciduria and galactosemia). The development of the disease can currently be prevented by correction (treatment) after the birth of the patient. Typical examples of such diseases are galactosemia, phenylketonuria, and hypothyroidism.
Accurate identification of the mutation in hereditary diseases is essential for their prenatal and preimplantation diagnosis. As a result of prenatal diagnosis, if mutations are detected in the fetus, the pregnancy is terminated, thereby excluding the birth of sick children. Diagnosing and identifying the carriage of mutations in parents allows patients to be offered preimplantation genetic diagnosis procedures when planning a pregnancy in order to exclude pathogenetically significant hereditary defects and, accordingly, reduce the risk of repeated birth of sick children in families, which should help reduce the burden of hereditary diseases and the costs for treatment and rehabilitation of patients.
SEARCH FOR MUTATIONS IN DNA
Advances in sequencing have made it possible to analyze the entire exome and genome; in practice, these approaches have become routine tools for the geneticist. These advances have led to significant improvements in diagnostic efficiency and an increase in the number of identifiable genes underlying rare diseases. One of the first studies in which exome sequencing was used to identify a causative mutation in a hereditary disease is the work of an international group of authors led by American researchers from the Howard Hughes Medical Institute [4]. Using exome sequencing, they established a molecular diagnosis in a child with suspected Bartter syndrome. According to the results of the analysis, a homozygous missense mutation in the gene SLC26A3 responsible for the development of congenital chloride diarrhea was identified. Thus, a diagnosis other than a referral was made, which was made possible by sequencing the entire genome coding sequence [4]. Since then, exome sequencing has been increasingly used in clinical practice and has significantly increased the efficiency of molecular diagnostics, shortening the “diagnostic odyssey” of patients with monogenic diseases.
Currently, despite significant progress in the development of technologies for the molecular genetic diagnosis of orphan diseases, many unresolved problems remain. The efficiency of detection of pathogenetically significant mutations using advanced technologies based on DNA analysis, such as exome and genomic sequencing, is estimated at 30 to 50% [5, 6]. Thus, a group of American scientists evaluated the diagnostic value of exome sequencing in children with monogenic diseases. In the analysis of 40 clinical cases, genetic defects were detected in 12 (30%) patients, among which 47% of mutations were not previously mentioned in the literature. In addition, 36 patients underwent an analysis of secondary findings in relation to the main diagnosis (“accidental” findings). As a result, in three patients (8%), genetic variants were identified that lead to disorders that require medical intervention [5].
Similar work was carried out by a group of Australian researchers that evaluated the diagnostic value of exome sequencing for children with hereditary diseases, while a molecular diagnosis was made in 52% of cases [6]. In addition, 35% of patients received diagnoses that differed from the referral one, and in 26%, the tactics of clinical management of the patient were corrected. The authors also performed an economic analysis of different patient diagnostic trajectories and found that exome analysis performed at initial presentation could result in an additional cost savings of AUD 9020 compared to standard monogenic disease diagnostic approaches.
One of the largest studies evaluating the effectiveness of exome sequencing analyzed 3040 patients. As a result, the overall diagnostic value of exome sequencing was 28.8%. It should be noted that, in the analysis of only probands, the diagnostic value was 23.6%, and in the analysis of three family members, it was 31% [7]. Thus, the trio analysis improves the efficiency of identifying the causative mutation by identifying genetic variants de novo, which simplifies the classification of new variants. It should be noted that the cost of research increases by a factor of three.
The use of whole genome sequencing, contrary to expectations of a significant increase in diagnostic efficiency, does not significantly improve the situation. As a result of whole genome sequencing of 103 patients from Canada with hereditary diseases, a molecular diagnosis was made in 41% of patients [8]. Nevertheless, the use of this approach made it possible to identify mutations in the noncoding DNA sequence in 18 patients. A meta-analysis conducted by Oxford researchers compared the cost and effectiveness of exome and genomic sequencing. A total of 27 studies using exome sequencing and three studies of whole genome sequencing were analyzed in the work. As a result, it was shown that, on average, the efficiency of exome sequencing was 35%, and that of genome sequencing was 49% [9].
In cases where it is not possible to identify pathogenic genetic variants, it is possible to reanalyze exome or genomic sequencing data after a certain time. In some cases, this makes it possible to identify variants that were not found in the first analysis. The increase in diagnostic value in this case is due to several factors [10]:
• discovery of new genes/variants associated with diseases;
• changing the classification of previously identified variants owing to the expansion of databases, conducting functional studies;
• improvement of reference genomes;
• development of bioinformatic algorithms for searching for variants, including using machine learning methods;
• analysis of new types of variants;
• collection of more detailed information about the patient and/or age-related changes in the patient’s clinical symptoms.
Thus, it is possible to increase the diagnostic value of exome/genomic sequencing by approximately 10–20% [10, 11], despite the fact that, in some cases, this approach does not allow the identification of new pathogenetically significant variants [12].
The efficiency of both exome and genome sequencing has its limitations. To a greater extent, this is due to the fact that techniques based on DNA analysis do not allow identification of mutations that do not affect the amino acid composition of the protein. Most of the pathogenetically significant genetic variants currently identified are missense and nonsense mutations (84%), since DNA sequencing was used to search for them. In addition, the efficiency of DNA sequencing is affected by problems with repetitive sequences, GC-rich regions, incomplete probe coverage of the coding sequence, and difficulties with short sequence alignment, leading to missed variants within poor coverage regions.
One of the mechanisms leading to the occurrence of hereditary diseases and not detected by DNA sequencing as changing the amino acid sequence of a protein is splicing disorder. At present, only variants affecting canonical splicing sites are taken into account when interpreting genomic data. The share of such genetic variants is estimated at about 8.7% [13]. At the same time, a group of researchers from Great Britain and Spain, using mathematical modeling, predicted that 62% of all pathogenetically significant genetic variants can lead to abnormalities in RNA splicing [14].
SPLICING IN NORM AND PATHOLOGY
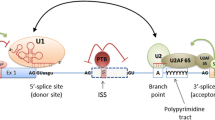
Currently, about 20 000 genes encoding human proteins and about 150 000 transcript isoforms are known. Therefore, on average, each human gene has about seven different isoforms [15]. Alternative splicing is characteristic of 90% of human intron-containing genes [16]. The main effector of the RNA splicing reaction is the spliceosome, a complex of hundreds of interacting proteins and small nuclear RNAs (snRNA), including small nuclear ribonucleoproteins (snRNP) U1, U2, U4, U5 and U6. Each pre-mRNA intron is flanked by a 5' exon and a 3' exon and contains various conserved splicing signals recognized by the spliceosome: 5' splicing site, branch point sequence, 3' splicing site, and polypyrimidine tract located 5–40 bp upstream of the 3' end of the intron. Since these splicing signals are insufficient to regulate splicing, the accuracy of pre-mRNA splicing depends on interactions between trans-acting factors (proteins and ribonucleoproteins) and cis-acting elements (pre-mRNA sequences), including exonic splicing enhancer, exonic splicing silencer, intronic enhancer splicing, and intronic splicing silencer. All of these elements exert their effects by modulating the binding of splicing factors, which in turn positively or negatively regulate the incorporation of a particular exon into the mature mRNA [17].
Pre-mRNA splicing plays an important role in the formation of protein diversity and the functioning of various cells and tissues of the body, which affects the role of disruption of normal splicing patterns in gene dysfunction and disease development. Diseases based on mutations affecting spliceosomes are described. Thus, mutations in the gene SNRPB, encoding the snRNP B and B1 polypeptides, lead to the development of cerebro-costo-mandibular syndrome; mutations in the gene EFTUD2 lead to the development of one of the types of mandibular-facial dysostosis; mutations in the gene SF3B4 (Splicing Factor 3b Subunit 4) have been identified in Nager syndrome; etc. [15]. Among the many genes responsible for the development of retinitis pigmentosa, six genes involved in pre-mRNA processing have been described (PRPF3, PRPF4, PRPF6, PRPF8, PRPF31, and SNRNP200) [18]. In addition, the role of alternative splicing in the development of solid malignant neoplasms has been shown [19–21]. Pathogenic genetic variants in genes SF3B1, U2AF1 and U2AF2 lead to the development of certain types of myeloid neoplasms [18].
Nevertheless, the main role of splicing in the development of various pathologies is due not to a violation of the mechanisms of pre-mRNA processing, but to changes in the regulatory sequences of the genes themselves. To search for such genetic variants, one of the convenient and affordable tools is RNA sequencing.
SEARCH FOR MUTATIONS USING RNA SEQUENCING
Single nucleotide variants in noncoding portions of genes may be responsible for much of the observed phenotypic variation [22]. Accurate pre-mRNA splicing required for proper protein translation depends on the presence of consensus sequences that define the boundaries between exons and introns and regulatory sequences recognized by the splicing mechanism. Point mutations in these consensus sequences can cause misrecognition of the exon and intron and lead to the formation of an aberrant gene transcript. Splicing mutation can occur in both introns and exons and disrupt existing splicing sites or splicing regulatory sequences, create new ones, or activate cryptic splicing sites. Typically, such mutations lead to errors in the splicing process and can lead to incorrect intron deletion, skipping, or the appearance of an extra exon [23].
To date, 23 868 mutations are known to lead to splicing disorders that are responsible for human hereditary diseases. The frequency of such genetic disorders is 8.7% of all mutations that cause hereditary diseases [13]. This number is probably an underestimate, since most of the described mutations were identified using genomic DNA sequencing without taking into account the effect of mutations on splicing. Recent studies point to the high frequency and important role of splicing mutations in the etiology of hereditary diseases, including Duchenne muscular dystrophy [24], cystic fibrosis [25], Ehlers–Danlos disease [26], hereditary diseases of the retina [27], and other monogenic pathologies. When analyzing genomic DNA, they can be easily overlooked and misclassified as synonymous changes or benign amino acid substitutions. However, RNA analysis clearly shows that such mutations have a significant effect on pre-mRNA splicing. It was assumed that larger genes with long introns were more prone to splicing defects, but it has now become clear that a significant number of mutations in smaller genes also cause abnormal mRNA splicing [28]. In addition, many of the identified splicing mutations are outside the canonical splicing sites and can be easily missed by genomic DNA analysis. There is growing evidence that misclassification of mutations is a common error, and the total number of splicing defects is likely to be underestimated [29].
In recent years, data have begun to accumulate on the use of RNA sequencing to search for pathogenetically significant mutations in patients with monogenic diseases (Table 1). According to some researchers, the diagnostic value of RNA sequencing is in the range of 10–35% for different groups of patients [30]. In addition, it has been shown that the use of RNA sequencing as a diagnostic tool can help expand our knowledge of the pathogenic significance of variants of unknown clinical significance (VUS) identified by DNA sequencing [17]. Table 1 provides information on studies to search for pathogenic genetic variants using RNA sequencing.
So, a group of researchers from Massachusetts analyzed 63 patients with suspected monogenic muscle diseases (myopathies and muscular dystrophies) and 184 control samples from the Genotype-Tissue Expression (GTEx) project. At the same time, 13 patients were diagnosed with pathogenic variants affecting the transcriptome (nonsense mutations and mutations in canonical splicing sites), which were used as a positive control. In 16 undiagnosed patients, predicted variants affecting splicing were identified by exome sequencing (n = 4), or strong candidate genes were identified (n = 12). In 34 patients, neither was identified. Muscle biopsy samples were used as material for the study. According to the results of the work carried out, pathogenic variants were identified in 35% of cases. The highest frequency of detection of pathogenic genetic variants missed by exome and whole genome sequencing was in the group of patients with predicted candidate variants (50%) and with a strong candidate gene (66%). At the same time, even in the group of patients without a candidate gene or candidate variant, mutations were detected in 21% of cases [30].
In a similar work by Canadian researchers on a sample of patients with neuromuscular diseases, more significant results were demonstrated in assessing the contribution of RNA sequencing to the search for new pathogenic mutations. RNA sequencing has been shown to detect the causative mutation in 36% of cases (9/25) in undiagnosed patients after exome sequencing [31].
Researchers from the Baylor College of Medicine (Houston, USA) performed transcriptome analysis on 182 patients with undiagnosed hereditary diseases after exome sequencing and chromosomal microarray analysis; as a result, pathogenic genetic variants were identified in 17% of cases [32]. Pathogenic mutations of various types were identified: mutations in canonical splice sites (7%), synonymous mutations in exons (7%), mutations in introns (43%), mutations in gene promoters (7%), and DNA copy number variations (36%).
Similar work was performed by S. Maddirevula et al., who performed whole transcriptome analysis of 155 patients without an identified mutation by exome sequencing. Transcript-deleterious variants (TDV) were found in 13.5% of cases. In addition, in this work, an analysis of tissue-specific gene expression with TDV was carried out. In particular, RNA samples obtained from blood, skin fibroblasts, and renal epithelial cells isolated from urine were analyzed. It was found that 84.1% (195 out of 232) of the analyzed genes are expressed in blood cell RNA, 85.8% (199 out of 232) in fibroblast RNA, and 90% (209 out of 232) in renal epithelial cell RNA. Most of the genes were expressed in all three RNA sources (75.5%), and only 2.6% (6 out of 232 genes) were not expressed in any of them [33].
The combination of DNA and RNA assessment methods leads to an increase in the diagnostic value of mass parallel sequencing, which was shown in the work of American researchers from the University of California. They analyzed 234 samples from undiagnosed patients using exome, whole genome, and transcriptome sequencing. As a result, the diagnostic value of DNA analysis methods was 31%, while the addition of RNA sequencing added another 7%, expanding the overall diagnostic value to 38%. In addition, 18% of the genetic variants identified by DNA sequencing were found to be pathogenic by RNA sequencing [34]. H. Wai et al. using quantitative real-time PCR and RNA sequencing analyzed the functional significance of 257 genetic variants with unclear clinical significance. As a result of the work carried out, it was found that 58 variants (33%) were associated with splicing anomalies; i.e., the pathogenetic significance of variants with unclear clinical significance was established [35].
One of the main difficulties in using RNA sequencing is the tissue-specific expression of many genes and the low or unavailability of many target tissues for analysis. While the most accessible material, peripheral blood, is used for analysis by DNA sequencing, this biological material can be uninformative for transcriptome analysis. A group of researchers from Stanford performed RNA sequencing from whole blood in 94 patients with suspected various orphan diseases (neurological, musculoskeletal and orthopedic, hematological and ophthalmic), but with an undetermined diagnosis. Expression in blood cells was shown for 76% of 284 genes associated with neurological disorders and for 66% of all genes sensitive to loss of function (loss-of-function intolerance). In 7.5% of cases, a diagnosis was made, with candidate genes identified for another 16.7%. The authors identified candidate genes using gene expression level analysis, allele-specific expression, and splicing abnormality prediction. This work showed the wide applicability of RNA sequencing technology, even for patients in whom the target tissue is difficult to access [36].
Another approach to search for splicing anomalies is to use computer programs and algorithms. Researchers at the Illumina Artificial Intelligence Lab developed the SpliceAI computer program to predict splicing anomalies in silico on the basis of DNA sequencing data [37]. “SpliceAI” is a residual neural network having a network architecture consisting of 32 extended convolutional layers that can recognize sequence determinants spanning large regions of the genome. To train the neural network, the authors used annotated pre-mRNA transcript sequences in GENCODE. At the same time, the accuracy of prediction of splicing events for pre-mRNA transcripts in the test data set was 95%. Even genes larger than 100 kb, such as CFTR, are often reconstructed with perfect accuracy [37]. Further improvement of approaches for finding splicing anomalies using simulation in silico can improve the efficiency of diagnosing hereditary diseases. In addition, this will expand our understanding of the mechanisms of regulation of such a complex process as pre-mRNA splicing.
Regardless of the approaches and methods used to search for splicing anomalies, the identified genetic variants require verification using functional analysis methods. The most convenient tool for this is the use of the minigene system. Minigenic constructs are sections of genes containing an exon and flanking intron regions with regulatory elements. The use of this model system makes it possible to determine the pathogenicity of various genetic variants by assessing their effect on splicing efficiency, as well as to search for exon and intron enhancers and splicing silencers. In addition, minigenes can be used to evaluate the role of splicing sites in establishing the baseline of exon recognition and to establish the role of various trans-regulators on individual splicing events [38].
RNA sequencing has good prospects as one of the tools for diagnosing hereditary orphan diseases. Nevertheless, there are a number of questions without answers to which it may be difficult to analyze and interpret the results obtained. These issues include tissue-specific gene expression and selection of tissues for analysis in various nosologies, optimization and standardization of the RNA sequencing technique and methods of bioinformatic data processing, understanding the required level of aberrant splicing for the occurrence of a pathological phenotype, and the influence of intragenic and intergenic contexts on the development of splicing anomalies. To answer all these questions, a more detailed study and deep understanding of the fundamental principles of pre-mRNA splicing is required. Knowledge of these mechanisms will not only improve diagnostics but also make significant progress in the treatment of orphan diseases with the help of drugs that modulate splicing.
REFERENCES
https://www.orpha.net/.
Nguengang Wakap, S., Lambert, D.M., Olry, A., et al., Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database, Eur. J. Hum. Genet., 2020, vol. 28, no. 2, pp. 165—173. https://doi.org/10.1038/s41431-019-0508-0
Online Mendelian Inheritance in Man. https://www.omim.org/.
Choi, M., Scholl, U.I., Ji, W., et al., Genetic diagnosis by whole exome capture and massively parallel DNA sequencing, Proc. Natl. Acad. Sci. U.S.A., 2009, vol. 106, no. 45, pp. 19096—19101. https://doi.org/10.1073/pnas.0910672106
Valencia, C.A., Husami, A., Holle, J., et al., Clinical impact and cost-effectiveness of whole exome sequencing as a diagnostic tool: a pediatric center’s experience, Front. Pediatr., 2015, vol. 3. https://doi.org/10.3389/fped.2015.00067
Tan, T.Y., Dillon, O.J., Stark, Z., et al., Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions, JAMA Pediatr., 2017, vol. 171, no. 9, p. 855. https://doi.org/10.1001/jamapediatrics.2017.1755
Retterer, K., Juusola, J., Cho, M.T., et al., Clinical application of whole-exome sequencing across clinical indications, Genet. Med., 2016, vol. 18, no. 7, pp. 696—704. https://doi.org/10.1038/gim.2015.148
Lionel, A.C., Costain, G., Monfared, N., et al., Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test, Genet. Med., 2018, vol. 20, no. 4, pp. 435—443. https://doi.org/10.1038/gim.2017.119
Schwarze, K., Buchanan, J., Taylor, J.C., et al., Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature, Genet. Med., 2018, vol. 20, no. 10, pp. 1122—1130. https://doi.org/10.1038/gim.2017.247
Robertson, A.J., Tan, N.B., Spurdle, A.B., et al., Reanalysis of genomic data: an overview of the mechanisms and complexities of clinical adoption, Genet. Med., 2022, vol. 24, no. 4, pp. 798—810. https://doi.org/10.1016/j.gim.2021.12.011
Liu, P., Meng, L., Normand, E.A., et al., Reanalysis of clinical exome sequencing data, N. Engl. J. Med., 2019, vol. 380, no. 25, pp. 2478—2480. https://doi.org/10.1056/NEJMc1812033
Tan, N.B., Stapleton, R., Stark, Z., et al., Evaluating systematic reanalysis of clinical genomic data in rare disease from single center experience and literature review, Mol. Genet. Genomic Med., 2020, vol. 8, no. 11, pp. 1—19. https://doi.org/10.1002/mgg3.1508
Stenson, P.D., Mort, M., Ball, E.V., et al., The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies, Hum. Genet., 2017, vol. 136, no. 6, pp. 665—677. https://doi.org/10.1007/s00439-017-1779-6
López-Bigas, N., Audit, B., Ouzounis, C., et al., Are splicing mutations the most frequent cause of hereditary disease?, FEBS Lett., 2005, vol. 579, no. 9, pp. 1900—1903. https://doi.org/10.1016/j.febslet.2005.02.047
Jiang, W. and Chen, L., Alternative splicing: human disease and quantitative analysis from high-throughput sequencing, Comput. Struct. Biotechnol. J., 2021, vol. 19, pp. 183—195. https://doi.org/10.1016/j.csbj.2020.12.009
Kalsotra, A. and Cooper, T.A., Functional consequences of developmentally regulated alternative splicing, Nat. Rev. Genet., 2011, vol. 12, no. 10, pp. 715—729. https://doi.org/10.1038/nrg3052
Marco-Puche, G., Lois, S., Benítez, J., et al., RNA-seq perspectives to improve clinical diagnosis, Front. Genet., 2019, vol. 10. https://doi.org/10.3389/fgene.2019.01152
Scotti, M.M. and Swanson, M.S., RNA mis-splicing in disease, Nat. Rev. Genet., 2016, vol. 17, no. 1, pp. 19—32. https://doi.org/10.1038/nrg.2015.3
Wu, Z.-H., Tang, Y., and Zhou, Y., Alternative splicing events implicated in carcinogenesis and prognosis of thyroid gland cancer, Sci. Rep., 2021, vol. 11, no. 1, p. 4841. https://doi.org/10.1038/s41598-021-84403-6
Marin, J.J.G., Reviejo, M., Soto, M., et al., Impact of alternative splicing variants on liver cancer biology, Cancers (Basel), 2022, vol. 14, no. 1, p. 18. https://doi.org/10.3390/cancers14010018
Kim, B.-H., Woo, T.-G., Kang, S.-M., et al., Splicing variants, protein—protein interactions, and drug targeting in Hutchinson—Gilford progeria syndrome and small cell lung cancer, Genes (Basel), 2022, vol. 13, no. 2, p. 165. https://doi.org/10.3390/genes13020165
Wachs, A.S. and Bohne, J., Two sides of the same medal: noncoding mutations reveal new pathological mechanisms and insights into the regulation of gene expression, WIREs RNA, 2021, vol. 12, no. 1, pp. 1—21. https://doi.org/10.1002/wrna.1616
Anna, A. and Monika, G., Splicing mutations in human genetic disorders: examples, detection, and confirmation, J. Appl. Genet., 2018, vol. 59, no. 3, pp. 253—268. https://doi.org/10.1007/s13353-018-0444-7
Habara, Y., Takeshima, Y., Awano, H., et al., In vitro splicing analysis showed that availability of a cryptic splice site is not a determinant for alternative splicing patterns caused by +1G>A mutations in introns of the dystrophin gene, J. Med. Genet., 2009, vol. 46, no. 8, pp. 542—547. https://doi.org/10.1136/jmg.2008.061259
Sanz, D.J., Hollywood, J.A., Scallan, M.F., et al., Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA, PLoS One, 2017, vol. 12, no. 9, p. e0184009. https://doi.org/10.1371/journal.pone.0184009
Symoens, S., Malfait, F., Vlummens, P., et al., A novel splice variant in the n-propeptide of COL5A1 causes an eds phenotype with severe kyphoscoliosis and eye involvement, PLoS One, 2011, vol. 6, no. 5. e20121. https://doi.org/10.1371/journal.pone.0020121
Weisschuh, N., Buena-Atienza, E., and Wissinger, B., Splicing mutations in inherited retinal diseases, Prog. Retin. Eye Res., 2021, vol. 80, p. 100874. https://doi.org/10.1016/j.preteyeres.2020.100874
Chen, M. and Manley, J.L., Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches, Nat. Rev. Mol. Cell Biol., 2009, vol. 10, no. 11, pp. 741—754. https://doi.org/10.1038/nrm2777
Xiong, H.Y., Alipanahi, B., Lee, L.J., et al., The human splicing code reveals new insights into the genetic determinants of disease, Science, 2015, vol. 347, no. 6218. https://doi.org/10.1126/science.1254806
Cummings, B.B., Marshall, J.L., Tukiainen, T., et al., Improving genetic diagnosis in Mendelian disease with transcriptome sequencing Genotype—Tissue Expression Consortium, Sci. Transl. Med., 2017, vol. 9, no. 386. https://doi.org/10.1126/scitranslmed.aal5209
Gonorazky, H.D., Naumenko, S., Ramani, A.K., et al., Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease, Am. J. Hum. Genet., 2019, vol. 104, no. 3, pp. 466—483. https://doi.org/10.1016/j.ajhg.2019.01.012
Murdock, D.R., Dai, H., Burrage, L.C., et al., Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing, J. Clin. Invest., 2021, vol. 131, no. 1. https://doi.org/10.1172/jci141500
Maddirevula, S., Kuwahara, H., Ewida, N., et al., Analysis of transcript-deleterious variants in Mendelian disorders: implications for RNA-based diagnostics, Genome Biol., 2020, vol. 21, no. 1, p. 145. https://doi.org/10.1186/s13059-020-02053-9
Lee, H., Huang, A.Y., Wang, L., et al., Diagnostic utility of transcriptome sequencing for rare Mendelian diseases, Genet. Med., 2020, vol. 22, no. 3, pp. 490—499. https://doi.org/10.1038/s41436-019-0672-1
Wai, H.A., Lord, J., Lyon, M., et al., Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance, Genet. Med., 2020, vol. 22, no. 6, pp. 1005—1014. https://doi.org/10.1038/s41436-020-0766-9
Frésard, L., Smail, C., Ferraro, N.M., et al., Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts, Nat. Med., 2019, vol. 25, no. 6, pp. 911—919. https://doi.org/10.1038/s41591-019-0457-8
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., et al., Predicting splicing from primary sequence with deep learning, Cell, 2019, vol. 176, no. 3, pp. 535—548. e24. https://doi.org/10.1016/j.cell.2018.12.015
Cooper, T.A., Use of minigene systems to dissect alternative splicing elements, Methods, 2005, vol. 37, no. 4, pp. 331—340. https://doi.org/10.1016/j.ymeth.2005.07.015
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (Federal Scientific and Technical Program for the Development of Genetic Technologies for 2019–2027, agreement no. 075-15-2021-1061, RF 193021X0029).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
This article does not contain any studies involving animals or human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Skryabin, N.A., Zhigalina, D.I. & Stepanov, V.A. The Role of Splicing in the Pathogenesis of Monogenic Diseases. Russ J Genet 58, 1208–1215 (2022). https://doi.org/10.1134/S1022795422100088
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1022795422100088