
Abstract
Hemophilia A is a frequent X-linked recessive blood clotting disorder. It is caused by mutations in the F8 gene (locus Xq28) and affects 1 in 5000 newborn boys. The aim of this study was to determine the spectrum of the F8 gene mutations in the Russian patients with hemophilia A. Samples from 117 unrelated families with an incoming diagnosis of hemophilia A were tested by IS-PCR, multiplex PCR, MPS technology, and quantitative MLPA analysis. Mutations were found in all 117 cases. Lof mutations in the VWF gene in the compound-heterozygous state were detected in two patients, two patients had pathogenic variants in the F9 gene, and one patient had a pathogenic variant in the F7 gene. These patients were excluded from further calculations. Intron 22 inversion was detected in 40% of cases, intron 1 inversion in 1% of cases, and gross deletions/duplications in 6% of cases. Point mutations accounted for 53%: missense mutations, 27%; small deletions, 10%; splice site mutations, 6%; nonsense mutations, 5%; and small duplications, 5%. Eighteen mutations were not described previously. Most of them are Lof mutations. Thus, it is necessary to employ different methods for the effective molecular diagnostics of hemophilia A.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Blood clotting is the process responsible for stopping bleeding in the case of vascular damage. It consists of a cascade of sequential activation of coagulation factors and results in formation of a clot. Each stage of this process may have disturbances associated with the absence or functional defect of one or several factors, which results in various coagulopathies. They include the hemophilia group. Hemophilia A is the most common form, with a frequency of 1 : 5000 newborn boys [1]. This disease is X-linked recessive. It is expressed as abundant, sometimes life-threatening bleeding, which can lead to disability. Depending on the activity of factor VIII (antihemophilic globulin), there are several degrees of disease severity: less than 1%, severe; 1–5%, moderate; 5–40%, mild (http://www.ehc.eu). Patients with severe hemophilia require regular intravenous administration of exogenous factor VIII.
Antihemophilic globulin is a multidomain protein synthesized in the endothelial cells of sinusoidal capillaries of the liver and in hepatocytes, from which it is secreted into the bloodstream. In its inactive state, factor VIII circulates in the blood, bound by the von Willebrand factor, which protects it from rapid degradation [2]. Factor VIII acts on the internal coagulation pathway of the blood, forming together with factor IX an internal tenase complex on the surface of activated platelets.
Factor VIII is encoded by the F8 gene. This is a large and complexly arranged gene consisting of 26 exons of 186 kb. The gene is localized in the telomeric region of the long arm of chromosome X in locus Xq28. It contains long intron sequences, where the size of some introns exceeds 14 kb.
In total, more than 3200 mutations in the F8 gene have been described in the HGMD database (http://www.hgmd.cf.ac.uk). They are evenly distributed throughout the gene and include both point changes in the sequence and gross structural changes. Two frequent mutations in the gene are described: intron 22 inversion and intron 1 inversion, in which patients with severe hemophilia account for 45–50 and 1–5%, respectively [3, 4]. These pathogenic variants are not found in patients with plasma factor VIII concentration of more than 2% [5].
Until recently, it was very difficult to search for mutations in the F8 gene and hemophilia A was diagnosed only indirectly. However, in some families, it was impossible to perform this study because of the absence of the patient’s biological material. In addition, the use of STR markers in prenatal diagnostics and diagnostics of carriage in female relatives of a patient has a number of risks, since often the markers used in indirect diagnostics are located not in the gene but at some distance from it, and in the case of chromosome X recombination in this locus, there is the likelihood of obtaining an erroneous result. There are also coagulopathies that are clinically and even biochemically disguised as hemophilia A, for example, von Willebrand disease type 2N or combined deficiency of factors V and VIII. These diseases have a different type of inheritance, which can lead to misinterpretation of the results of indirect diagnostics.
With the development of new methods, in particular, IS-PCR (inverse shifting polymerase chain reaction), the method of quantitative MLPA analysis (multiplex ligation-dependent probe amplification), and the MPS method (massive parallel sequencing), it became possible to diagnose hemophilia A directly, revealing the F8 gene mutation in the majority of burdened families in patients and female carriers, thus eliminating all the risks associated with indirect diagnostics.
The purpose of this work is to study the range of mutations in Russian patients with hemophilia A.
MATERIALS AND METHODS
For this study, 117 families with the incoming diagnosis “hemophilia A” were selected from the archive of the DNA diagnostics laboratory of the Research Centre for Medical Genetics. In 79 families, the patient’s DNA sample was suitable for analysis, while in another 38 families the study was carried out using the material of patient’s female relatives who were carriers of this disease according to the results of previously carried out indirect DNA diagnostics or genealogical analysis.
DNA isolation in patients and their relatives from whole blood collected in a test tube with an EDTA anticoagulant was carried out using the Wizard® Genomic DNA Purification Kit (Promega, United States) according to the manufacturer’s protocol.
The search for inversions of intron 22 and intron 1 of the F8 gene was carried out using the IS-PCR and extended IS-PCR methods according to the protocols proposed in the works of L.C. Rossetti et al. [6, 7].
A search for small deletions and insertions of the F8 gene as well as gross deletions (in patients) was carried out using the multiplex PCR system developed in the DNA diagnostics laboratory of the Research Centre for Medical Genetics, which includes the coding regions of all 26 exons of this gene (Table 1). Amplification of the studied DNA fragments multiplexes 1–4 was carried out using an MS2 programmable thermal cycler produced by DNA-Technology (Russia) in a reaction mixture volume of 25 μL of the following composition: 0.1–0.2 μg genomic DNA, 0.4 pM each original oligoprimer, 0.2 mM each nucleoside triphosphate, 4 mM MgCl2, 1.0 activity unit Biotaq DNA polymerase (BioMaster), 1× PCR buffer (67 mM Tris-HCl, 16.6 mM (NH4)2SO4, 0.01% Twin-20, pH 8.8), and 20–30 μL mineral oil.
PCR was carried out in the following mode: initial denaturation at 95°C for 5 min; then 30 cycles of temperature change: denaturation at 94°C for 45 s, annealing of primers at 62°C for 45 s, chain elongation at 72°C for 45 s; and final elongation at 72°С for 7 min.
A 7% polyacrylamide gel with an acrylamide : bisacrylamide ratio of 29 : 1 was used to evaluate the amplification results. After separation of the fragments, the gel was stained in a solution of ethidium bromide (0.1 μg/mL in 1× TBE) for 10 min and washed with water. Electrophoresis results were visualized using the Bio-Rad (United States) GelDoc documenting system in UV radiation with a wavelength of 312 nm. The results were interpreted on the basis of the lengths of the fragments identified by electrophoresis compared with the control samples.
The analysis of point mutations of the F8, F9, and VWF genes was carried out using the MPS method on a next generation sequencer Ion S5 using the custom panel Coagulum, which includes the coding sequences of these genes. The size of the panel is 28 150 bp. According to the data of AmpliSeq™ Сoverage Analysis, its calculated coverage is 95.7%. Ultramultiplex PCR combined with subsequent sequencing (AmpliSeq™) was used for sample preparation. The sequencing results were processed using the standard automated algorithm proposed by Thermo Fisher Scientific (Torrent Suite™), as well as the Gene-Talk software program (http://www.gene-talk). The depth of reading of all the identified variants was at least 200×. The dbSNP (https://www.ncbi.nlm.nih.gov/ projects/SNP/) and the Genome Aggregation databases (http://gnomad.broadinstitute.org/) were used to estimate the population frequencies of the identified variants. The pathogenicity of the variants was estimated according to the guidelines for the interpretation of data obtained by mass parallel sequencing methods [8].
The number of copies of all 26 exons of the F8 gene was determined by quantitative MLPA analysis using the SALSA MLPA P178 F8 probemix kit (MRC Holland, Amsterdam, Netherlands) according to the manufacturer’s protocol. The separation of the obtained fragments was carried out using a 3130 ABI Genetic Analyzer device for capillary electrophoresis (Applied Biosystems, United States). The results were estimated using the Coffalyser program (MRC Holland, Netherlands) (https://www.mlpa.com).
In one family, a clinical exome was studied on a next generation IlluminaNextSeq 500 sequencer using the method of pair-terminal reading (2 × 75 bp). For sample preparation, we used the technique for selective capture of DNA regions belonging to the coding regions of the 6277 genes currently described as clinically significant (SeqCap EZ HyperCap Workflow kit). Processing of the sequencing data was carried out using the standard automated algorithm proposed by Illumina for data analysis at https://basespace.illumina.com.
The nucleotide variants detected by the multiplex PCR system and the MPS method were verified using Sanger sequencing on the 3130 ABI Genetic Analyzer device for capillary electrophoresis (Applied Biosystems, United States).
RESULTS AND DISCUSSION
Since the most frequent pathogenic variants in patients with severe hemophilia A are major F8 gene rearrangements, it is optimal to start diagnosing this disease by searching for inversions of intron 22 and intron 1. For the detection of these mutations, the most widely used method is IS-PCR.
In the sample under study, the intron 22 inversion was detected in 45 families and the intron 1 inversion was detected in one patient, which constitutes 38% and slightly less than 1% of cases, respectively (Fig. 1). The frequencies of these inversions in Russian patients hardly differ from those in other countries [3–5, 9].

Electrophoregram of the results of detection of inversions of introns 1 and 22 (Inv1 and Inv22) of the F8 gene using the IS-PCR method. (1) Molecular weight marker λ/Pst1; (2) Inv22 in the heterozygous state; (3) Inv1 in the hemizygous state; (4, 6, 9) the norm; (5) a fragment of anomalous length in the heterozygous state was detected in the patient’s daughter; then the family was found to have deletion of exons 15–21; (7, 8) a fragment of anomalous length was detected in the proband and his mother in the hemi- and heterozygous state, respectively; then the family was found to have deletion of exons 15–21; (10) the proband has no band corresponding to the norm or no inversion of intron 1; then the family was found to have duplication of exons 2–13; (11) abnormal Inv22 in the hemizygous state (no band corresponding to the norm or no intron 22 inversion, then abnormal Inv22 was detected by the extended IS-PCR method); (12) Inv22 in the hemizygous state. Vertical arrows indicate fragments with altered electrophoretic mobility or missing fragments.
Among families with intron 22 inversion, besides classical inversions detected by IS-PCR, one case of abnormal inversion was observed, which was manifested in the absence of electrophoretic fragments corresponding to normal or inverted intron 22 (Fig. 1). Further, using the extended IS-PCR method, it was found that the intron 22 inversion in this family was combined with an unspecified rearrangement of the noncoding intergenic region (data not shown).
In addition, in another three families, the IS-PCR method revealed abnormal patterns that were characterized by the absence or change in the length of the amplified fragments that would correspond to the norm or inversions of intron 22 and 1 (Fig. 1). Additional research methods (multiplex PCR system and quantitative MLPA analysis) in these families revealed the presence of gross structural changes: deletions of exons 15–21 in two families and duplications of exons 2–13 in one family. Such complex structural rearrangements of the F8 gene have already been described by other researchers. In this case, presumably, an inversion occurs at the first stage of such a rearrangement, leading to instability of this region, which causes further deletions and duplications of different exons of the F8 gene or insertions of other genes into a given locus [10–12]. The reason for such frequent structural anomalies of the F8 gene is the fact that its very long introns contain a large number of various repeats and homologous regions, which contributes to the occurrence of gross rearrangements during meiosis. Moreover, the probability of inversion is 10 times higher in male germ cells and the probability of deletion is 5 times higher in female cells [13, 14].
Thus, the total informativity of the IS-PCR method, taking into account gross deletions confirmed by quantitative MLPA analysis, was 42%. The fact that, in addition to the classical inversions of introns 1 and 22 of the F8 gene, this method also detected anomalous variants showed that this method has broader diagnostic capabilities and allows detecting some gross structural rearrangements.
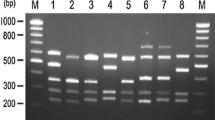
In 68 families with the incoming diagnosis of hemophilia A in which no inversions were detected, it was necessary to further search for point mutations of the F8 gene. The most effective method in this case is the method of mass parallel sequencing. However, because of the high cost and complexity of the method, these patients were first tested using the simple and fast multiplex PCR system that includes the coding regions of all 26 exons of the F8 gene. This system makes it possible to identify small deletions, insertions, some nucleotide substitutions, and also gross deletions (in male probands). According to global data, the total share of small deletions, insertions, and gross deletions in the F8 gene accounts for about 20% of mutations [9]. In this study, multiplex PCR detected 16 fragments with altered electrophoretic mobility, in which Sanger sequencing later detected point pathogenic variants (Table 2, Fig. 2) and one gross deletion containing the sequence of exon 14. The total informativity of the multiplex PCR system taking into account the two gross deletions detected previously by IS-PCR was 16%.

Electrophoregram of the results of detecting some pathogenic variants of the F8 gene by multiplex PCR. (1, 7, 9, 11, 14) the norm; (6, 10, 13, 16) deletion of exons 15–21 in the hemizygous state; (2) c.3409_3410delCT in the hemizygous state; (3) c.2514_2526del13 in the heterozygous state; (4) c.411_412delСА in the hemizygous state; (5) c.214G>A (p.E72K) in the hemizygous state; (8) c.1844_1861del18 in the heterozygous state; (12) c.6906delT in the heterozygous state; (15) c.96dupA in the heterozygous state. The arrows indicate fragments with altered electrophoretic mobility or missing fragments.
Next, 51 families without the identified mutation were studied by mass parallel sequencing using a custom panel that includes the coding sequences of genes F8, F9, and VWF. Mutations were identified in 48 cases: in 44 families, pathogenic variants were found in gene F8 (Table 3), in two probands in gene F9, and in another two probands in the compound-heterozygous state in gene VWF (Table 4). The total informativity of the MPS method taking into account the point mutations and gross deletions previously identified by the multiplex PCR system was 57%.
Three families in which mutations using the approaches applied were not identified were studied by quantitative MLPA analysis. In two cases, gross duplications of gene F8 were found: duplications of exons 1–22 in one family and a complex structural rearrangement accompanied by duplication of exons 11–14 and 19–26 in another family. Thus, in total, three gross deletions and three gross duplications were observed in the sample under study. The informativity of the method of quantitative MLPA analysis for the unexamined sample of patients with the incoming diagnosis of hemophilia A was 5%.
In one family in which no mutations in the genes studied were detected at the final stage of the molecular genetic survey, a search for pathogenic variants was carried out using the method of clinical exome sequencing. The study was carried out using the material of the proband’s mother, since the patient’s biological material was unsuitable for this analysis. As a result, the F7 gene was found to contain the described pathogenic variant c.1061C>T (p.A354V) in the heterozygous state (Table 4). Probably, the disease in this family was caused by the deficit of factor VII. However, for the unequivocal assertion of this fact, it is necessary to identify the second mutation in the patient.
Thus, using the algorithm applied to examine families with the incoming diagnosis of hemophilia A, mutations were detected in all 117 families. The aggregate informativity of all the methods applied was 100%. F8 gene mutations were detected in 95.7% of cases, F9 gene mutations in 1.7%, VWF gene mutations in 1.7%, and F7 gene mutations in 0.9% of cases.
This level of informativity corresponds to world data, the value of which currently reaches 99% [15]. With the development of technology, the proportion of patients with hemophilia A for whom the mutation is not identified is decreasing. In the literature, the absence of pathogenic variants in such patients is explained by the fact that mutations can be located in regulatory regions or deep in introns [16, 17]. These variants can be detected only by expensive analysis of the genome; however, the interpretation of the clinical significance of such variants is very complicated, since it is difficult to explain the mechanism by which the identified change in the nucleotide sequence affects the structure of the protein encoded by this gene. The absence of the mutation in mothers of probands may also be due to the occurrence of a pathogenic variant in germ cells de novo, or mothers may have mosaicism with a very low proportion of the clone carrying the pathogenic variant. In addition, mutations in other genes leading to a phenotype similar to hemophilia A have been described. For example, there is a combined deficiency of factors V and VIII, which is due to the absence or functional defect of intracellular protein transporters of these factors encoded by genes MCFD2 and LMAN1 [18], or von Willebrand disease type 2N caused by such mutations in the VWF gene in which the binding of factor VIII to the von Willebrand factor is disturbed [19].
The shares of different types of mutations in the F8 gene in the samples examined were as follows: Inv22, 40%; missense mutations, 27%; small deletions, 10%; splice site mutations, 6%; nonsense mutations and small duplications, 5% each; gross deletions and duplications, 3% each; Inv1, 1% (Fig. 3). The vast majority in these samples are mutations leading to a severe course of hemophilia A, since they significantly disrupt the structure and, therefore, the functional activity of antihemophilic globulin. The distribution of the shares of different types of mutations in general corresponds to those described in the literature for patients with severe hemophilia A (Fig. 3). Therefore, it can be assumed that our sample mostly included patients with the severe form of the disease.

The fractional distribution of mutations in the F8 gene in the sample of patients with the incoming diagnosis of hemophilia A in this study and patients with severe hemophilia A in the meta-analysis of S.C. Gouw [9].
Almost all the identified point variants of the F8 gene are unique and were detected in the sample one time. Only transversion c.362G>T (p.G121V) and transition c.5122C>T (p.R1708C) were observed two and three times, respectively. While the transversion detected twice can be considered as a random event, transition c.5122C>T is probably a frequently repeated variant, since its share in the Factor VIII Gene (F8) Variant Database (http: //www.factorviii-db.org) accounts for about 0.6% of all cases of the disease.
The study revealed 18 pathogenic variants not described previously (Table 5). Most of them are Lof (lost of function) mutations. Of particular interest are the newly discovered missense variants p.I585K and p.G638C. In these positions, other amino acid substitutions, p.I585R and p.I585T and p.G638D, p.G638S, and p.G638V, respectively, have already been described as pathogenic. All previously described variants were registered in patients with severe hemophilia A, however, the amino acid substitution p.I585T was also found in patients with moderate severity of the disease (http://www.factorviii-db.org). Both codons are located in domain A2, which is responsible for the binding of activated factor IX. Therefore, any changes in the spatial conformation of the domain may disturb the indicated function to a certain extent. In the two missense mutations identified in this study, new amino acids differ from amino acids in the normal protein chain in size, charge, and hydrophobicity, which affects the structure of the protein. Thus, neutral isoleucine in position 585, located in the binding site of factor IX, is replaced by a larger positively charged lysine. This leads to its repulsion from neighboring amino acid residues, which changes the conformation of the domain and disrupts the interaction with factor IX [20, 21]. The replacement of glycine located in the depth of domain A2 by larger cytosine at position 638 can also change the structure of the domain and disturb the binding of factors VIII and IX.
Thus, the investigation of families with the incoming diagnosis of hemophilia A in probands revealed patients with mutations not only in the F8 gene but also in the genes of the VII, IX, and von Willebrand factors. This fact confirms the difficulty of correctly diagnosing a patient at the clinical examination stage. Only molecular genetic analysis allows differential diagnosis of clinically similar diseases. Establishing the type of mutation also makes it possible to predict the risk of such a serious complication in a patient with hemophilia A as the production of antibodies to exogenous factor VIII. It is known that the highest risk is in patients with gross deletions (up to 75%) and the lowest one is in patients with missense mutations (9%) [22].
In this study, a full molecular genetic survey of the sample of Russian patients with the incoming diagnosis of hemophilia A was carried out for the first time. Despite the diversity of the types of mutations that were found in the gene of factor VIII and the fact that the sample included patients with other clinically similar coagulopathies, owing to a combination of different methods, pathogenic variants were identified in all the families examined. The largest share in the F8 gene was made up of mutations leading to a severe course of the disease. The results of the study showed that, without the use of molecular genetic methods, differential diagnostics of various types of coagulopathies is sometimes difficult. Identifying the cause of the disease is extremely important for direct pre-implantation or prenatal diagnostics in the family. The study of the molecular genetic grounds of hemophilia A allows one to predict the risk of complications in a patient and select the optimal treatment and also contributes to the development of new therapy technologies.
REFERENCES
Mannucci, P.M. and Tuddenham, E.G., The hemophilias—from royal genes to gene therapy, N. Engl. J. Med., 2001, vol. 344, no. 23, pp. 1773—1779. https://doi.org/10.1056/nejm200106073442307
Orlova, N.A., Kovnir, S.V., Vorobiev, I.I., et al., Blood clotting factor VIII: from evolution to therapy, Acta Nat., 2013, vol. 5, no. 2, pp. 19—39.
Schroder, J., El-Maarri, O., Schwaab, R., et al., Factor VIII intron-1 inversion: frequency and inhibitor prevalence, J Thromb. Haemost., 2006, vol. 4, no. 5, pp. 1141—1143. https://doi.org/10.1111/j.1538-7836.2006.01884.x
Bagnall, R.D., Waseem, N., Green, P.M., and Giannelli, F., Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A, Blood, 2002, vol. 99, no. 1, pp. 168—174.
Kumar, P., Faridi, N.J., Husain, N., et al., Study of intron 22 inversion mutation in north India with review, Blood Coagul. Fibrinolysis, 2013, vol. 24, no. 2, pp. 120—124. https://doi.org/10.1097/MBC.0b013e3283574f40
Rossetti, L.C., Radic, C.P., Larripa, I.B., and De Brasi, C.D., Genotyping the hemophilia inversion hotspot by use of inverse PCR, Clin. Chem., 2005, vol. 51, no. 7, pp. 1154—1158. https://doi.org/10.1373/clinchem.2004.046490
Rossetti, L.C., Radic, C.P., Larripa, I.B., and De Brasi, C.D., Developing a new generation of tests for genotyping hemophilia-causative rearrangements involving int22h and int1h hotspots in the factor VIII gene, J. Thromb. Haemost., 2008, vol. 6, no. 5, pp. 830—836. https://doi.org/10.1111/j.1538-7836.2008.02926.x
Ryzhkova, O.P., Kardymon, O.L., Prokhorchuk, E.B., et al., Guidelines for the interpretation of massive parallel sequencing variants (MPS), Med. Genet., 2017, no. 7, pp. 4—17.
Gouw, S.C., Berg, H.M., Oldenburg, J., et al., F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis, Blood, 2012, vol. 119, no. 12, pp. 2922—2934. https://doi.org/10.1182/blood-2011-09-379453
Fujita, J., Miyawaki, Y., Suzuki, A., et al., A possible mechanism for Inv22-related F8 large deletions in severe hemophilia A patients with high responding factor VIII inhibitors, J. Thromb. Haemost., 2012, vol. 10, no. 10, pp. 2099—2107. https://doi.org/10.1111/j.1538-7836.2012.04897.x
Sanna, V., Ceglia, C., Tarsitano, M., et al., Aberrant F8 gene intron 1 inversion with concomitant duplication and deletion in a severe hemophilia A patient from Southern Italy, J. Thromb. Haemost., 2013, vol. 11, no. 1, pp. 195—197. https://doi.org/10.1111/jth.12061
Zimmermann, M.A., Oldenburg, J., Muller, C.R., and Rost, S., Unusual genomic rearrangements in introns 1 and 22 of the F8 gene, Hamostaseologie, 2011, vol. 31, suppl. 1, pp. S69—S73.
Becker, J., Schwaab, R., Moller-Taube, A., et al., Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type-dependent sex ratio of mutation frequencies, Am. J. Hum. Genet., 1996, vol. 58, no. 4, pp. 657—670.
Rossiter, J.P., Young, M., Kimberland, M.L., et al., Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells, Hum. Mol. Genet., 1994, vol. 3, no. 7, pp. 1035—1039.
Bastida, J.M., Gonzalez-Porras, J.R., Jimenez, C., et al., Application of a molecular diagnostic algorithm for haemophilia A and B using next-generation sequencing of entire F8, F9 and VWF genes, Thromb. Haemost., 2017, vol. 117, no. 1, pp. 66—74. https://doi.org/10.1160/th16-05-0375
Pezeshkpoor, B., Zimmer, N., Marquardt, N., et al., Deep intronic “mutations” cause hemophilia A: application of next generation sequencing in patients without detectable mutation in F8 cDNA, J. Thromb. Haemost., 2013, vol. 11, no. 9, pp. 1679—1687. https://doi.org/10.1111/jth.12339
Inaba, H., Shinozawa, K., Amano, K., and Fukutake, K., Identification of deep intronic individual variants in patients with hemophilia A by next-generation sequencing of the whole factor VIII gene, Res. Pract. Thromb. Haemost., 2017, vol. 1, no. 2, pp. 264—274. https://doi.org/10.1002/rth2.12031
Zhang, B., McGee, B., Yamaoka, J.S., et al., Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2, Blood, 2006, vol. 107, no. 5, pp. 1903—1907. https://doi.org/10.1182/blood-2005-09-3620
Casonato, A., Galletta, E., Sarolo, L., and Daidone, V., Type 2N von Willebrand disease: characterization and diagnostic difficulties, Haemophilia, 2018, vol. 24, no. 1, pp. 134—140. https://doi.org/10.1111/hae.13366
Plantier, J.L., Saboulard, D., Pellequer, J.L., et al., Functional mapping of the A2 domain from human factor VIII, Thromb. Haemost., 2012, vol. 107, no. 2, pp. 315—327. https://doi.org/10.1160/th11-07-0492
Venkateswarlu, D., Structural insights into the interaction of blood coagulation co-factor VIIIa with factor IXa: a computational protein-protein docking and molecular dynamics refinement study, Biochem. Biophys. Res. Commun., 2014, vol. 452, no. 3, pp. 408—414. https://doi.org/10.1016/j.bbrc.2014.08.078
Oldenburg, J. and Pavlova, A., Genetic risk factors for inhibitors to factors VIII and IX, Haemophilia, 2006, vol. 12, suppl. 6, pp. 15—22. https://doi.org/10.1111/j.1365-2516.2006.01361.x
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest. The authors declare that they have no conflict of interest.
Statement of compliance with standards of research involving humans as subjects. All procedures carried out in this study with the participation of people comply with the ethical standards of the institutional and/or national research ethics committee and the 1964 Helsinki Declaration and its subsequent changes or comparable standards of ethics. All patients or their legal representatives and family members provided voluntary informed consent for the processing and publication of molecular genetic examination data on conditions of anonymity. The scientific theme in the framework of which this work was carried out was approved by the ethics committee of the Research Centre for Medical Genetics.
Additional information
Translated by K. Lazarev
Rights and permissions
About this article

Cite this article
Beskorovainaya, T.S., Milovidova, T.B., Schagina, O.A. et al. Complex Molecular Diagnostics of Hemophilia A in Russian Patients. Russ J Genet 55, 1015–1024 (2019). https://doi.org/10.1134/S1022795419080027
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1022795419080027